>Corresponding Author : Mourad Boukheloua
>Article Type : Case Report
>Volume : 3 | Issue : 5
>Received Date : 08 Aug, 2023
>Accepted Date : 21 Aug, 2023
>Published Date : 26 Aug, 2023
>DOI : https://doi.org/10.54289/JCRMH2300123
>Citation : Boukheloua M. (2023) Biventricular Arrhythmogenic Cardiomyopathy with Delayed Revelation: A Case Report. J Case Rep Med Hist 3(5): doi https://doi.org/10.54289/JCRMH2300123
>Copyright : © 2023 Boukheloua M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access
Associate Professor, Cardiology Department, UHC Hussein-Dey. Cardiology Department, Pr Nafissa Hamoud Hospital, CHU Hussein-Dey, Algiers, Algeria
*Corresponding author: Boukheloua M, Associate Professor, Cardiology Department, UHC Hussein-Dey. Cardiology Department, Pr Nafissa Hamoud Hospital, CHU Hussein-Dey, Algiers, Algeria
Abstract
Arrhythmogenic right ventricular dysplasia (ARVD), currently known as arrhythmogenic right-predominant cardiomyopathy (ARVC), is a cardiomyopathy that affects the right ventricle preferentially, but may also involve the left ventricle or be biventricular; with a variable clinical course ranging from asymptomatic cases to sudden cardiac death, which may be inaugural.
Research to date has tended to focus on young patients in their thirties, but here we present the case of a patient presenting with syncope of rhythmic origin, diagnosed with biventricular ARVC at the age of 65 years old, illustrating the difficulty of the diagnostic and therapeutic management of this pathology, because of the variability of clinical presentation, incomplete penetrance, lack of specificity of diagnostic criteria, and low sensitivity of imaging techniques ; diagnosis is made on the basis of a cluster of arguments established by the ESC 2010 Task-Force, or on the more recently proposed, but as yet unvalidated, Padua criteria, including cardiac MRI.
Thus, although ARVD is a rare hereditary disease, it must be considered even in advanced age, and appropriately managed with medical treatment and devices to prevent sudden cardiac death, as recommended by ESC 2022.
Keywords: Arrhythmogenic RV Dysplasia; Biventricular Involvement; Sudden Death; Implantable Cardioverter Defibrillator
Abbreviations: nEHRA: European Heart Rhythm Society, HRS: Heart Rhythm Society, ICD: Implantable Cardioverter Defibrillator, NSVT: Non-Sustained Ventricular Tachycardia, PA/VC : Premature Atrial/Ventricular Contraction PL/SAX: Parasternal Long/Short Axis, R/LV: Right/Left Ventricle, ARVD: Arrhythmogenic Right Ventricular Dysplasia, ARVC: Arrhythmogenic Right-Predominant Cardiomyopathy
Introduction
Arrhythmogenic right ventricular dysplasia (ARVD) was described in the late 1970s by Fontaine and Marcus under the term arrhythmogenic right ventricular dysplasia (ARVD), a term which is still sometimes used, although it is gradually being replaced by the term predominantly right arrhythmogenic cardiomyopathy [1], this is a cardiomyopathy that affects the right ventricle preferentially, but may also involve the left ventricle or be biventricular in very rare cases reported in the literature to date ; characterized genetically by desmosome abnormalities, pathophysiologically by fibroadipous infiltration replacing the ventricular myocardium, and clinically by variability ranging from asymptomatic cases to syncope secondary to ventricular arrhythmias, even leading to sudden death [2,3,4,5], recent data have demonstrated the value of ICD in primary or secondary prevention. Research to date has tended to focus on young patients in their early thirties, and here we present the case of a patient presenting with syncope from a rhythmic origin, diagnosed with biventricular DAVD at the age of 65.
Case presentation
A 65-year-old patient with no previous history of syncopal episodes presented in the emergency department, with ventricular tachycardia reduced by external electric shock. During observation, the patient presented a rhythmic storm; the various electrocardiograms recorded showed polymorphic ventricular tachycardias, when the EKG was in sinus rhythm, microvoltage with negative T waves in the right precordial leads were noted (figure 1); 24-hour Holter EKG recording revealed biomorphic, few short-coupled ESAs with the presence of polymorphic and few long-coupled 358/24-hour PVCs, isolated and sometimes trigeminal with the presence of several 42/24-hour doublets, with no intra- or interventricular conductive disturbances or pauses (figure 2); transthoracic echocardiography revealed tetra-cavity dilatation with 30% impaired left ventricular systolic function, right ventricular dysfunction and generalized hypokinesia with multiple intra-LV thrombi of different ages. Given that ischemic etiology is most common at this age, we completed our investigations with coronary angiography, which doesn't revealed any significant stenosis ; a cardiac MRI was then performed, showing dilatation of the RV infundibulum, measured at 40 mm, the site of marked dyskinesia; the free wall of the RV was indented and pseudoaneurysmal, particularly in its anterior and inferior parts; severe hypokinesia of the lateral wall of the LV; multiple bi-ventricular mesocardial contrasts, involving the lateral wall of the LV and quasi-transmural in the anteroseptal wall of the LV, and transmural in the wall of the infundibulum of the LV as well as in its inferior apical part, in late enhancement after gadolinium injection (figure 3); the diagnosis of arrhythmogenic dysplasia with biventricular involvement was therefore retained.
The patient was treated for heart failure, at the same time, Amiodarone was introduced as an antiarrhythmic agent with effective anticoagulation; after an echocardiographic check-up confirming the regression of thrombi, our patient was implanted with a cardiac defibrillator for secondary prevention. His assessment three months after implantation showed an asymptomatic patient with improved left ventricular function (LVEF: 40%), with five appropriate shocks from the ICD, illustrating the value of this management according to ESC 2022 recommendations.
Discussion
Arrhythmogenic cardiomyopathy is predominantly right ventricular, but can also affect the left ventricle or be biventricular, as in the case of the patient we have just described [1]; while the average age of diagnosis is around thirty, with extremes ranging from the second to the fourth decade, and is much more common in adolescents and young adults, in rare cases such as the one presented here, revelation may be delayed due to the variability of the clinical presentation depending on the stage and age of onset, with a natural history ranging from strictly asymptomatic early forms, with the risk of sudden death, particularly during physical activity, through the electrical phase characterized by the presence of palpitations or syncope in connection with ventricular arrhythmias linked to structural anomalies affecting the LV and/or RV, to the phase of right or left ventricular dysfunction, depending on which ventricle is affected, with progressive dilatation and loss of contractility leading to heart failure. This is a genetic disease in which a desmosomal protein abnormality, transmitted in an autosomal dominant mode with variable penetrance, is at the origin of the fibro-adipocytic infiltration of the ventricular myocardium [2,3].
However, a number of limitations must be taken into account in the diagnosis of ARVC, which remains difficult because of the variability of clinical presentation, incomplete penetrance, lack of specificity of diagnostic criteria, and the low sensitivity of imaging techniques for diagnosis. As a result, the diagnosis is established on the basis of a set of arguments grouped together by the ESC 2010 Task-Force [3], or on the Padua criteria proposed more recently, which have not yet been validated [4] (based on careful analysis of a 12-lead ECG, high-amplification ECG, 24-hour Holter ECG, stress ECG for diagnostic purposes in triggering ventricular arrhythmias, or for prognostic purposes in monitoring therapeutic efficacy), In addition, 2D echocardiography can be used to exclude differential diagnoses, but may return without abnormality, particularly in the early stages, without ruling out the diagnosis, and cardiac MRI with late enhancement - recently included in the Padua criteria - in connection with fibrosis, considered to be the arrhythmogenic substrate).
In 2020, a panel of experts identified limitations to the reference diagnostic criteria of the ESC 2010 Task Force: errors in interpreting ECG or imaging results, presentations similar to other diseases such as sarcoidosis, and the inclusion of genetic tests in the diagnostic criteria lead to overdiagnosis, on the other hand, the exclusion of tissue characterization criteria on cardiac MRI in the 2010 ESC Task-Force reference diagnostic criteria has led to underdiagnosis [4], but as experience has been gained and MRI techniques have advanced, MRI has become an indispensable tool in the diagnosis of ARVC [5].
It is essential to stratify rhythmic risk, according to the 2019 HRS recommendations on ARVC, major rhythmic risk factors are syncope, NSVT, positive SVP and LV dysfunction with LVEF <50%, minor factors are male gender, ESV load>1000/24h, LV dysfunction, propositus status and the presence of multiple pathogenic variants [5].
Management is symptomatic, palliative rather than curative, first and foremost through a healthy lifestyle in which the practice of sports should be limited, since it could aggravate or accelerate the progression of the disease. The EHRA expert consensus recommends avoiding competitive sports or activities of moderate to high intensity [6], and anticoagulation has been initiated for secondary prevention in accordance with the HRS 2019 recommendations, but also because of symptomatic recurrent VT as antiarrhythmic therapy in addition to Cordarone, since it has been shown to be the most effective combination with the aim of reducing the incidence of arrhythmias and/or the number of appropriate shocks after ICD implantation, which is retained as secondary prevention in our patient according to HRS 2019 recommendations [5]; ARVC often has an impact on patients' quality of life, due to symptoms, treatment side-effects and the psychological repercussions of the disease. Psychological care should therefore not be neglected; regular monitoring in consultation is essential, and certain examinations may need to be repeated (ECG, Holter ECG, stress test, cardiac ultrasound every 12 to 24 months and cardiac MRI every three to five years) to adapt treatment ; the appearance of any new symptom should be reported, and cardiac monitoring of all first-degree relatives should be undertaken from the age of ten onwards. At the same time, genetic counseling should be offered initially on a case-index basis, since the genetic origin of the disease, the mode of transmission and the benefits of family cardiological monitoring should be explained [7].
Conclusion
Although ARVC is a rare hereditary disease, it must be diagnosed even at an advanced age, with reference to the criteria of the ESC 2010 Task Force, which are still valid, and then managed appropriately with medical treatment and devices to prevent sudden cardiac death, as codified by ESC 2022. The case we have just presented illustrates the difficulty of the diagnostic and therapeutic management of this particular disease, which remains a challenge due to the variability of its presentation and evolution.
Attachments
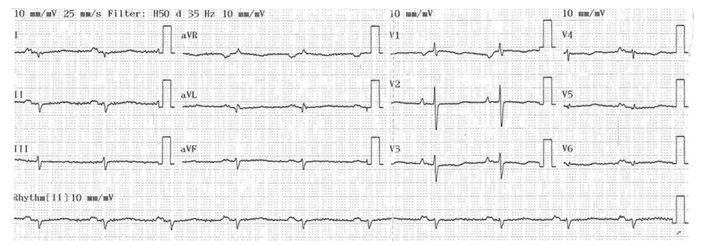
Figure 1: Electrocardiogram: areas of fibrosis lead to parietal conductive and repolarization disorders, such as negative T waves in precordial leads and microvoltage in advanced disease.
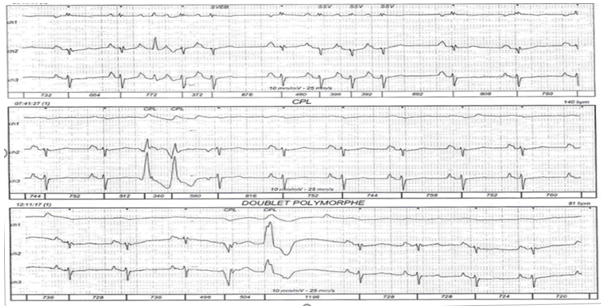
Figure 2: 24-hour ECG Holter: ESA salves, ESV doublets.
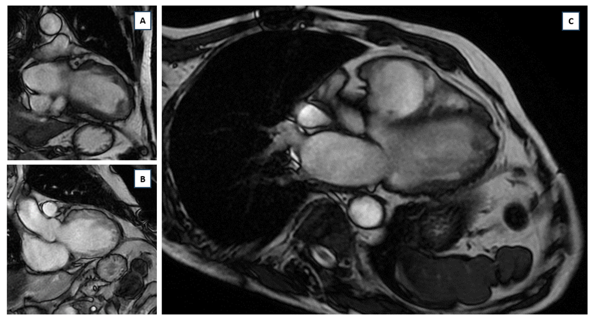
Figure 3: MRI images showing: (A): intra-LV thrombus, (B): dilatation of the infundibulum of the RV measured at 40mm, site of marked dyskinesia; pseudoaneurysmal scalloped free wall of the RV, particularly in its anterior and inferior parts; (C): dilatation with multiple bi-ventricular, mesocardial contrast, involving the lateral wall of the LV and almost transmural in the anteroseptal wall of the LV, and transmural in the wall of the infundibulum of the LV as well as in its infero-apical part, with late enhancement time after gadolinium injection.
Bibliography
- Marcus FI, Fontaine GH, Guiraudon G, et al. (1982) Right ventricular dysplasia: a report of 24 adult cases. Circulation. 65: 384-398. [PubMed.]
- Azaouagh A, Churzidse S, Konorza T, Erbel R. (2011) Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. Clin Res Cardiol. 100(5): 383-394. [Ref.]
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, et al. (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur Heart J. 31: 806-814. [Ref.]
- Corrado D, Marra MP, Zorzi A, Beffagna G, Cipriani A, et al. (2020) Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 319: 106-114. [PubMed.]
- Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, et al. (2019) HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 16(11): e301-e372. [Ref.]
- Ruwald AC, Marcus F, Estes NA, Link M, McNitt S, et al. (2015) Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 36(27): 1735-1743. [PubMed.]
- Sen-Chowdhry S, Syrris P, McKenna WJ. Role of Genetic Analysis in the Management of Patients With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J Am Coll Cardiol. 50(19): 1813-1821. [Ref.]