>Corresponding Author : Tabat Meryem
>Article Type : Case Report
>Volume : 3 | Issue : 6
>Received Date : 04 Sep, 2023
>Accepted Date : 20 Sep, 2023
>Published Date : 26 Sep, 2023
>DOI : https://doi.org/10.54289/JCRMH2300128
>Citation : Meryem T, Karim M, Marouane S, Zineb EJ, Haboub M, et al. (2023) A Rare Case of Non-Ischemic Dilated Cardiomyopathy Revealing Inclusion Body Myositis. J Case Rep Med Hist 3(6): doi https://doi.org/10.54289/JCRMH2300128
>Copyright : © 2023 Meryem T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Case Report | Open Access
Department of Cardiology. Ibn Rochd University Hospital of Casablanca, Morocco
*Corresponding author: Tabat Meryem, Department of Cardiology. Ibn Rochd University Hospital of Casablanca, Morocco
Abstract
Inclusion body myositis is the most common inflammatory myopathy after age 50.
Unlike polymyositis and dermatomyositis, in which cardiac involvement is relatively common, current evidence indicates that inclusion body myositis is not associated with cardiac disease such as dilated cardiomyopathy. We report the observation of a 57-year-old patient known to be type 2 diabetic under treatment, admitted with acute congestive heart failure. the paraclinical examinations carried out were in favor of inclusion body myositis complicated by heart failure and dilated cardiomyopathy, the patient was treated with boluses of corticosteroids and immunosuppressants as well as treatment of heart failure, this attitude has improved his symptoms as well as his quality of life.
The interest of this work is to underline the importance of diagnosis and early treatment in the management of myositis complicated by cardiovascular manifestations, particularly dilated cardiomyopathy.
Keywords: Inclusion Body Myositis; Dilated Cardiomyopathy; Acute Decompensated Heart Failure; Inflammation
Abbreviations: DCM: Dilated cardiomyopathy
Introduction
Inclusion body myositis is the most common inflammatory myopathy after age 50; a prevalence of between 9.3 and 14.9 per million has been found in Western countries [1]. Although the etiology is still poorly understood, a range of examinations can guide the diagnosis, in particular the histological examination of muscle biopsies shows the coexistence of inflammatory and muscular degenerative aspects [2]. This myopathy is characterized by asymmetrical muscle weakness (quadriceps weakness, finger weakness, feet dropping or dysphagia) [3]. Direct cardiac involvement has so far not been demonstrated, we report a case of cardiomyopathy dilated in a 57-year-old patient in whom no apparent explanation for cardiac dilation was found.
Observation
We report the case of a 57-year-old patient, known to have type 2 diabetes for 10 years on oral antidiabetics. The patient was initially admitted to the cardiology intensive care unit for predominantly left-sided congestive heart failure decompensated by a respiratory infection. The clinical examination found a conscious patient, hemodynamically stable, normotensive at 119/65 mmhg, tachycard at 110 beats/min, dyspneic with the presence of jugular vein turgor, edema of the lower limbs, crackling rales mid- fields. the neurological examination revealed a motor deficit in all 4 limbs, 3/5 proximally and 2/5 distally. The electrocardiogram showed a regular sinus rhythm at 105 beats/min, a normal heart axis, QRS at 110 ms, without secondary repolarization disorder. Chest x-ray revealed cardiomegaly, bilateral interstitial syndrome, and right basithoracic focus. A transthoracic echocardiography showed a dilated left ventricle, with an end-diastolic diameter of (58mm, indexed at 37mm/m2) (figure 1), global hypokinesia, severe left ventricular systolic dysfunction, with an ejection fraction of the left ventricle at 30%, moderate mitral regurgitation, left atrial dilatation, pulmonary arterial hypertension at 43 mmhg (figure 2), dilated lower vena cava not very compliant.
In view of the clinical symptoms, a dosage of muscle enzymes: creatine kinase and LDH were elevated (470UI/L and 640UI/L) respectively. Thus, an electromyogram was carried out objectifying a myogenic trace, these data raised the suspicion of myositis complicated by dilated cardiomyopathy hence the carrying out of a dosage of auto antibodies: anti-synthetase and antiSRP ac came back positive as well as a muscle biopsy which showed a severe variation in the size of fibers and inflammatory infiltrates with focal invasion of the muscle.
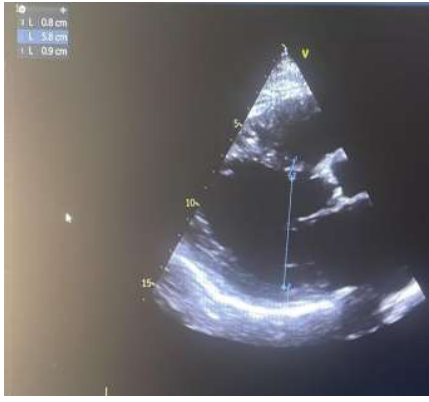
Figure 1: A transthoracic echocardiography showed a dilated left ventricle.
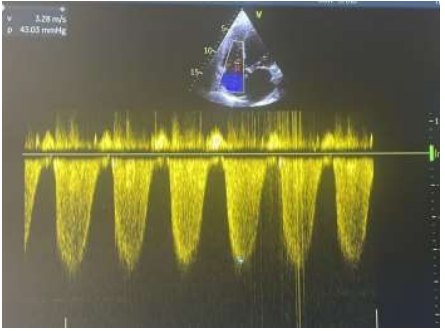
Figure 2: Pulmonary arterial hypertension at 43 mmhg
The initial treatment consisted of setting conditions, oxygen therapy, initiation of medical treatment, based on intravenous loop diuretics, and potassium sparing (spirinolactone), inhibitors of converting enzyme (ramipril), antibiotic therapy based on amoxicillin, clavulanic acid with control of his diabetes, beta-blockers were introduced after stabilization.
Coronary angiography showed normal coronary arteries (figure 3). Extensive investigations have shown no findings supporting endocrine, metabolic, infectious, or toxic etiologies of cardiomyopathy.
The patient was transferred after stabilization to the neurology department where he received corticosteroid boluses, immunosuppressants and motor physiotherapy sessions.
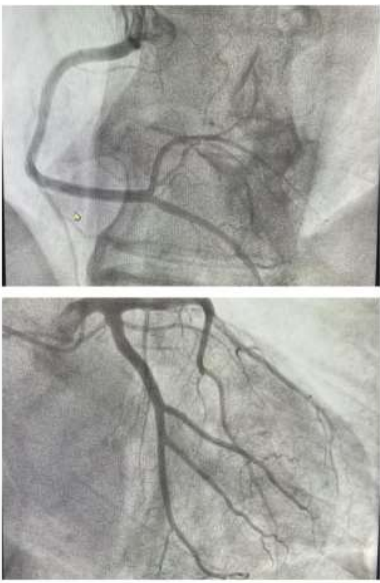
Figure 3: A coronary angiography without significant lesions
Discussion
Inclusion body myositis is the most common subtype of autoimmune myopathy in patients older than 50 years. Diagnostic criteria were proposed in 2011 by the European Neuromuscular Center, these criteria are based on clinical and paraclinical data in particular the histological results of muscle biopsy, have a high specificity greater than 99%, but their sensitivity is low at 57%.[4].
Dilated cardiomyopathy (DCM) is one of the cardiomyopathies, a group of diseases that primarily affect the myocardium. It has different causes and is classically classified into ischemic and non-ischemic dilated cardiomyopathy. Non-ischemic dilated cardiomyopathy has diverse and varied etiologies (infectious, metabolic, genetic diseases, etc.) [5].
Cardiovascular manifestations have been reported in patients with dermatomyositis or polymyositis, the most commonly reported being congestive heart failure, conduction abnormalities, which can lead to complete heart block, and coronary artery disease [6]. While the data concerning cardiac damage in patients with inclusion body myositis are still rare. In a study conducted in the Netherlands aimed at exploring the prevalence and nature of cardiac abnormalities in inclusion body myositis, fifty one patients underwent transthoracic echocardiography, the results were as follows: systolic dysfunction in 8%, ventricular hypertrophy in 27%, and diastolic dysfunction in 45 patients [7]. Thus in another Swedish study, they carried out measurements of troponin T (cTnT) in 42 patients suffering from inclusion body myositis, 26 patients (62%) had a cTnT level > 0.05 microg/L, the threshold used for the diagnosis of myocardial infarction.
Patients on immunosuppressive treatment had lower cTnT levels than those not treated, so none of the patients showed signs of myocardial damage or renal failure at the time of sampling [8]. In this patient, there were no cardiovascular risk factors, nor physiopathological causes explaining the dilation and dysfunction of the LV which were found by in-depth investigations in particular: a coronary angiography, an endocrine and metabolic assessment made of: (thyroid assessment, calcium, phosphoremia, zincemia), viral serologies (HIV, HVB, HVC, Syphilis) as well as a toxic and immunological investigation, all this exhaustive assessment returned normal. Cardiac involvement leads to a poorer prognosis in patients with inclusion body myositis; therefore, due to the rarity of the disease and the lack of randomized controlled trials, there is currently no specific treatment for cardiac complications, current recommendations are based solely on reports and case series [9].
Our patients were placed on immunosuppressants, corticosteroids and heart failure medications aimed at preventing deterioration of LV systolic function, reducing major cardiovascular events and improving cardiac remodeling quality of life.
To our knowledge, dilated cardiomyopathy was only described in very rare cases. It should be noted that the association between inclusion body myositis and dilated cardiomyopathy observed in our patient may be a coincidence, however this observation may suggest the hypothesis of an association between the two conditions.
Conclusion
This case presentation shows the importance of thinking about myositis as a cause of nonischemic dilated cardiomyopathy. Such a diagnosis is associated with a poor prognosis with a risk of cardiovascular death and hospitalization for heart failure.
The use of immunosuppressive agents and heart failure medications improves left ventricular systolic and diastolic function, cardiac symptoms and quality of life.
Authors' contributions:
Tabat Meryem: patient management, data collection and analysis.
Karim Mounaouir: paper preparation
Salmaoui Marouane and El Jaouhari Zineb: manuscript writing and revision.
All the authors have read and agreed to the final manuscript.
Competing interests: The authors declare no competing interests.
References
- Needham M, Corbett A, Day T, Christiansen F, Fabian V, et al. (2008) Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci. 15(12): 1350-1353. [PubMed.]
- Nelke C, Kleefeld F, Preusse C, Ruck T, Stenzel W. (2022) Inclusion body myositis and associated diseases: An argument for shared immune pathologies. Acta Neuropathol Commun. 10(1): 84. [PubMed.]
- Needham M, James I, Corbett A, Day T, Christiansen F, et al. (2008) Sporadic inclusion body myositis: phenotypic variability and influence of HLA-DR3 in a cohort of 57 Australian cases. J Neurol Neurosurg Psychiatry. 79(9): 1056-1060. [PubMed.]
- Panginikkod S, Musa R. (2023) Inclusion Body Myositis. In: StatPearls Treasure Island (FL): StatPearls Publishing. PMID: 30855788. [PubMed.]
- Mahmaljy H, Yelamanchili VS, Singhal M. (2023) Dilated Cardiomyopathy. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. PMID: 28722940. [PubMed.]
- Lundberg IE. (2006) The heart in dermatomyositis and polymyositis. Rheumatology (Oxford). 45(Suppl 4): iv18-21. [PubMed.]
- Cox FM, Delgado V, Verschuuren JJ, Ballieux BE, Bax JJ, et al. (2010) The heart in sporadic inclusion body myositis: A study in 51 patients. J Neurol. 257(3): 447-451. [Ref.]
- Lindberg C, Klintberg L, Oldfors A. (2006) Raised troponin T in inclusion body myositis is common and serum levels are persistent over time. Neuromuscul Disord. 16(8): 495-497. [Ref.]
- Jayakumar D, Zhang R, Wasserman A, Ash J. (2019) Cardiac Manifestations in Idiopathic Inflammatory Myopathies: An Overview. Cardiol Rev. 27(3): 131-137. [PubMed.]