>Corresponding Author : Nikolaos Andreas Chrysanthakopoulos
>Article Type : Review Article
>Volume : 3 | Issue : 7
>Received Date : 30 Sep, 2023
>Accepted Date : 13 Oct, 2023
>Published Date : 17 Oct, 2023
>DOI : https://doi.org/10.54289/JCRMH2300135
>Citation : Chrysanthakopoulos NA and Vryzaki E. (2023) The Special Role of JAK/STAT, and Notch Signaling Pathways in Cancer Pathogenesis. J Case Rep Med Hist 3(7): doi https://doi.org/10.54289/JCRMH2300135
>Copyright : © 2023 Chrysanthakopoulos NA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Review Article | Open Access
1Dental Surgeon, Oncologist, PhD in Oncology, Specialized in Clinical Oncology, Cytology and Histopathology, Dept. of Pathological Anatomy, Medical School, University of Athens, Greece Resident in Maxillofacial and Oral Surgery, 401 General Military Hospital of Athens, Greece Consultant in Dentistry, NHS of Greece, Athens, Greece
2Department of Dermatology, Rio University Hospital of Patras, Greece
*Corresponding author: Nikolaos Andreas Chrysanthakopoulos, Dental Surgeon, Oncologist, PhD in Oncology, Specialized in Clinical Oncology, Cytology and Histopathology, Dept. of Pathological Anatomy, Medical School, University of Athens, Greece Resident in Maxillofacial and Oral Surgery, 401 General Military Hospital of Athens, Greece Consultant in Dentistry, NHS of Greece, Athens, Greece
Abstract
Cancer is essentially a genetic disease, characterized by unclear etiology in many cases, complex pathogenic mechanisms, since in the majority of cancer cases only risk or predisposing factors have been recognized, and unclarified observations concerning organs that are not often affected by malignant tumors such as the small intestine, spleen, heart, and the neurons of the Central Nervous System. A tumor’s appearance and progression presupposes a series of genetic and non-genetic alterations that characterize tumor cells, and are related to self-sufficiency in growth stimuli, insensitivity to anti-growth signals, unlimited replicative potential, angiogenesis promotion, ability to infiltrate surrounding tissues and induce distant metastases, and resistance to apoptosis. The mentioned characteristics are associated with signaling pathways that are involved in cell survival, cell death, cell growth and division, cell motility, and can be considered in the context of abnormalities of wider signaling networks that support cancer progression, such as tumor microenvironment alterations, angiogenesis, and inflammation. Among others, two signaling pathways, JAK/STAT and Notch, seem to play essential roles in tumorigenesis, despite the fact that are implicated in diverse cellular functions such as tissue formation and cell differentiation, proliferation, apoptosis, inflammation, self-renewal, antitumor immune response suppression, motility, stress response and other responses depending on the target tissue. Hyperactivation of those signaling pathways caused by mutations are able to transform cellular proto-oncogenes to oncogenes, whereas inactivation of tumor suppressor genes eradicates crucial negative regulators of signaling. Various anticancer agents target those signaling pathways in an attempt to inhibit those pathways including monoclonal antibodies and other agents in the context of targeted cancer therapy.
Keywords: Cancer; Cancer Pathogenesis; Cellular Signaling Pathways
Abbreviations: VEGF: Vascular Endothelial Growth Factor, PDGF: Platelet-Derived Growth Factor, EGF: Epidermal Growth Factor, MYN: Myelohyperplastic Neoplasms, ITH: Idiopathic Thrombocytosis, PMF: Primary Myelofibrosis, GLL: Granulocytic Lymphocytic Leukemia, HNSCC: Head And Neck Squamous Cell Carcinoma, DLBCL: Diffuse Large B-Cell Lymphoma, JMML: Juvenile Myelomonocytic Leukemia, T-ALL: T-Cell Acute Lymphoblastic Leukemia, BC: Breast Cancer, LA: Lung Adenocarcinoma, HCC: Hepatocellular Carcinoma, OC: Ovarian Cancer, CRC: Colorectal Cancer, CLL: Chronic Lymphocytic Leukemia, ACC: Adenoid Cystic Carcinoma, MCL: Mantle Cell Lymphoma, SMZL: Splenic Marginal Zone Lymphoma, PC: Prostate Cancer, CCRCC: Clear Cell Renal Cell Carcinoma, Cscc: Cutaneous Squamous Cell Cancer, Pdac: Pancreatic Ductal Carcinoma, Panin: Pancreatic Intraepithelial Neoplasia, EPO: Erythropoietin, G-CSF: Granulocyte Colony-Stimulating Factor, GM-CSF: Granulocyte-Macrophage Colony-Stimulating Factor, LIF: Leukemia Inhibitory Factor, Jaks: Janus Kinases, SCID: Severe Combined Immunodeficiency, PIAS: Protein Inhibitor Of Activated Signal Transducer And Activator Of Transcription, Cdks: Cyclin-Dependent Kinases, EMT: Epithelial-Mesenchymal Transition, MMP: Matrix Metallo-Proteinase, AR: Androgen Receptor, CLS: Cytoplasmic Localization Signal, TGF-Α: Transforming Growth Factor-Alpha, SCLC: Small Cell Lung Cancer,OPN: Osteopontin, IHC: Immune-Histochemical, GC: Gastric Carcinoma, Gics: Glioma-Initiating Cells, Tams: Tumor-Associated Macrophages, SCC: Squamous Cell Carcinoma, NSCLC: Non-Small Cell Lung Cancer, NASH: Non-Alcoholic Steatohepatitis, IGF: Like Growth Factor, NET: Neuroendocrine Tumors
Introduction
Cell signaling pathways constitute an extremely complex network, that connect and interact with each other, are activated following the effect of various extracellular growth factors such as Vascular Endothelial Growth Factor (VEGF), Platelet-Derived Growth Factor (PDGF), Epidermal Growth Factor (EGF), etc. and intracellular molecules that act as classical oncogenes, such as myc, fos, jun [1,2]. The extracellular factors that activate similar receptors in the cell membrane actually transport information from neighboring cells and from the extracellular matrix and affecting the intracellular ones, regulate cellular processes involving in development, differentiation, polarity, motility, position change, proteins production as well as apoptosis - programmed cell death. The mentioned cellular processes differ according to the cell type, the cellular microenvironment, the interactions with neighboring cells and the extracellular matrix. The exact mode of operation of those pathways with the characteristic complex networks and interactions of those pathways have not fully elucidated and this is one of the reasons that make it difficult to be effective treatment of most types of malignant tumors [2]. The complicated structure and complexity of those pathways has important implications for the understanding of cancer cell behavior and therefore the possibility of using this knowledge in targeted cancer therapies. Biological processes such as cell survival, development, differentiation, motility, proliferation of cells, etc. are regulated by multiple signaling pathways and the alterations that are observed in cancer cells are the effect of multiple changes and interactions in them [3].
The JAK/STAT signaling pathway is responsible for embryonic development regulation and is implicated in the control of procedures such as maintenance of stem cell, hematopoiesis, inflammatory reaction, and transduces signals from cytokines and growth factors that act through a trans-membrane receptor families series [4]. STAT dimers bind specific promoter sequences and are responsible for the regulation of genes transcription that control functions such as cell differentiation, proliferation, apoptosis [5], inflammation, self-renewal, antitumor immune response suppression, motility, stress response and other responses depending on the target tissue [6] (Figure 1). The JAK/STAT pathway has also influences on gene expression outside the “normal” signaling cascade of phosphorylation, contributing to chromatin epigenetic modifications [7], as it has been observed that activated JAK2 is removed in nucleus and modify histones although that observation remains controversial [7,8]. Several members of the JAK/STAT family have been involved in diverse malignancies. The JAK/STAT activation has been extensively investigated for its role in hematologic malignancies [9] and provides the basis for its investigation in solid tumors, whereas it contributes to the acquirement of two critical cancer hallmarks, invasion and metastasis. It has been found that 50-95% of patients with classic myelohyperplastic neoplasms (MYN), idiopathic thrombocytosis (ITH), polycythemia vera (CP), and primary myelofibrosis (PMF), had an activating mutation in JAK2 (a valine to phenylalanine change, V617F) in the malignant clone [10]. Activating mutations that concern STATs, although it is a rare event, have been observed in cancer. In granulocytic lymphocytic leukemia (GLL) cases, a percentage of 40% had mutations that affect the STAT3 SH2 domain which introduce hydrophobic residues that are considered to stabilize STAT dimers, and result in increased STAT-responsive transcription [11]. STAT5A/B locus amplification has been revealed in prostate cancer cases and was associated with STAT5 increased expression and its nuclear localization in tumor samples [12]. STAT3 is a functional signaling protein, its constitutive activation has been involved in the inflammation progression and inflammation-associated cancers, whereas is expressed in many cells and tissues, and plays a critical role in the cell proliferation, regulation, survival, differentiation, and apoptosis. It is highly expressed in tumor cells, due to the loss of precise negative regulation and persistent activation. It also shows antiapoptotic effect [13]. Persistent STAT3 activation is closely associated with hyperproliferation and anti-apoptosis in malignant cells [14,15]. STAT3 is also constitutively activated in tumors, such as in colon, lung, liver, breast gland, pancreas, prostate, stomach, brain, ovaries, leukemia and in lymphoma cases, head and neck squamous cell carcinoma (SCC), melanoma, kidney, and esophageal cancer [16-18]. STAT5 signaling plays a role in proliferation and invasion in glioblastoma cases [19]. In general, STAT3 and STAT5 A/B promote tumorigenesis, whereas STAT1 activation has opposite effects [20]. Although unusual, JAK1 mutations affect many hematological malignancies. To be more specific, the JAK2V617F, JAK1V658F JAK1 homolog, results in the kinase constitutive activation [21]. In a study examined 186 adult leukemia samples, 4 JAK1 mutations were found, of which 75% were JAK1V658F [22], whereas JAK1 mutations were observed in 4 out of 45 T-cell prolymphocytic leukemia patients, including one patient harboring JAK1V658F [10]. In diffuse large B-cell lymphoma (DLBCL) has been recorded epigenetic gene regulation by JAK1 [23]. A wide variety of JAK3 mutations have been detected in many cell lines and patient samples, and the most widely observed mutations concerned those within the pseudokinase domain, and were associated with several lymphoma and leukemia cases, including T-cell adult lymphocytic leukemia (7%) [24], natural killer T-cell lymphoma (21%-35%) [25,26], juvenile myelomonocytic leukemia (JMML)(~10%) [27], and T-cell prolymphocytic leukemia (32%-42%) [10,28]. Mutations within the kinase domain are associated with JMML (10-40%) [29]. JAK2-mediated chromatin modification has been revealed in primary mediastinal and Hodgkin lymphoma, both of which share common molecular and biological characteristics, as autocrine IL-13 production and chromosome 9p24 amplification both result to JAK2 activation [30,31].
The Notch pathway is developmentally a highly conserved pathway implicated in morphogenesis, tissue formation and cell differentiation. It is also involved in several malignancies and can behave as an oncogene or a tumor suppressor gene, depending on the cell type, whereas it is very important for cellular functions in different tissues, such as skin, CNS, pancreas, muscle and hematopoietic system [32-34]. In most cases the Notch pathway inhibits the initial differentiation of the cell causing a second differentiation or maintains the cell in an undifferentiated condition. It has been characterized as "the guard" against differentiation [32]. Under normal circumstances Notch signaling is essential for the development and homeostasis of normal intestinal epithelial cells, as regulates the colonic goblet cells and stem cells differentiation [35,36]. The Notch pathway is one of the most important pathways in embryonic development. Since tumorigenesis and organ growth share the same mechanisms it is not surprising that growth-related pathways such as Notch, Hedgehog and Wnt play a role in cancer cell growth and proliferation. Highly aggressive cancer cells have many features of embryonic progenitor and use the Notch pathway for their survival. Notch pathway dysfunction is associated with many types of cancer and as mentioned, can have an oncogenic or tumor suppressor effect depending on the tissue or organ it is expressed. However, it remains unknown how the same pathway activation leads to two opposing actions in different cell types. A possible explanation for that paradoxical behavior is that Notch activates/deactivates specific genes depending on the cell or pathways that determine the final effect of Notch pathway on the cell [37]. It was the first detected as an oncogene in T-cell acute lymphoblastic leukemia (T-ALL) cases [38,39], and the Notch receptors alteration was identified in several cancer types (Table 1). Thus, Notch activation in breast cancer (BC), lung adenocarcinoma (LA), hepatocellular carcinoma (HCC), ovarian cancer (OC), colorectal cancer (CRC), and leukemia was defined to be oncogenic [40-43] (Table 1). The Notch activation pattern varies, as it can be activated by upstream signals or by structural alteration because of its internal mutations. Possible tumorigenesis mechanisms concern control of the tumor-initiating cell phenotype, regulation of known upstream or downstream tumor-associated signaling molecules, such as p53 or myc, promotion of tumor invasion or angiogenesis, cell cycle regulation, etc. [44].
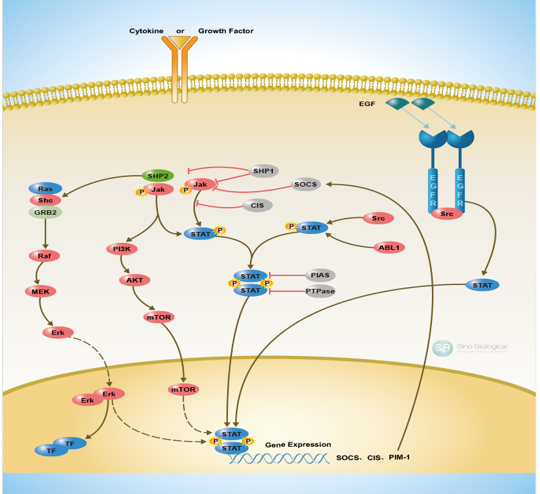
Figure 1. JAK/STAT signaling involvement in cellular functions and other signaling pathways
Notch receptors mutations result in Notch receptor gain-of-function (e. g. in hematological malignancies [45,46]) or loss-of-function (e. g. in bladder cancer [47] and squamous cell carcinomas [48], gastric and other gastrointestinal cancers [49]) (Table 1) and are major causes of Notch signaling dysregulation. Similarly, Notch pathway hypo- and hyperactivation can lead to tumorigenesis, depending on the tissue type, the type of receptor-ligand interactions and the genetic alteration. Notch1 mutations, for instance, have been revealed in HNSCC, esophagus and skin and have been associated with the pathway hypoactivation, whereas, Notch1 hyperactivation has been associated with the etiology of T-ALL and chronic lymphocytic leukemia (CLL), BC, adenoid cystic carcinoma (ACC) and mantle cell lymphoma (MCL) [38,39,49]. A proportion greater than 50% of T-ALL cases had Notch1 somatic activating mutations [50], and a rate almost 58% of splenic marginal zone lymphoma (SMZL) cases showed activating Notch mutations, known as NNK-SMZLs [51]. Notch signaling was considered oncogenic in glioma cases, in which it maintains brain cancer stem cells [52]. In a medulloblastoma mouse model [53] Notch1 had potentially oncogenic effects in the brain in association with other oncogenic hits, such as p53 loss. The Cancer Genome Atlas Research Network suggested that approximately 23% of OC patients had Notch signaling alterations [54], whereas similar researches showed that Notch1 and-3 promote OC development [54-56]. The Jagged1 distribution and expression were found to be associated with prostate cancer (PC) [57,58]. A higher Notch1and Notch3 expression has been detected [59,60] in NSCLC, and in LA initiation and progression [61-63] suggesting that Notch acts as an oncogene, and that alteration implicated NUMB expression loss, that increases Notch activity, and Notch1 gene [64] gain-of-function mutations. ACC of salivary gland, frequently is characterized by activating Notch1 and Notch2 mutations [65-67], as Notch1 inhibitors showed notable antitumor efficacy [68]. An overexpression of Notch ligands and receptors have been found in clear cell renal cell carcinoma (CCRCC) cases [69]. Notch receptors were also frequently mutated, in cutaneous squamous cell cancer (cSCC) and its adjacent normal tissue, leading to down-regulation or loss of function [70]. Similarly, Notch1 and Notch2 malfunction were detected in lung SCC cases [71].
Recent studies have revealed that proliferation and apoptotic events can be influenced by Notch1 signaling. In cancer cases such as PC, ACC, and BC activated Notch1 repressed p27 to promote cell cycle and malignant proliferation [72-75].
Notch may be implicated in many cancer types as a pro-tumor effector, however it can also act as a tumor suppressor in others, such as SCC and neuroendocrine tumors [76] (Table 2). A negative association between Notch and carcinogenesis was also observed in bladder [77], esophageal [78,79], and cervical SCC cases [80]. Inactivated Notch1-3 has been observed in SCC specimens, [81,82], such as in 40% of HNSCC cases that concerned inactivated Notch1 [83,84]. Notch mutation is frequent in pancreatic ductal carcinoma (PDAc) [85], as Notch1 is able to inhibit the pancreatic intraepithelial neoplasia (PanIN) formation in a PDAC mouse model [86]. In addition, Notch1 loss is a necessary process in a Kras-induced PDAC mouse model [87], suggesting its role as a tumor suppressor gene. However, previous surveys suggested that Notch shows an oncogenic role in PDAC development [88-90]. Notch signaling is activated in PDAC, that causes the premalignant PDAC cells development [88].
The purpose of the current review was the presentation of the latest as possible approaches to cellular signaling pathways implicated in cancer pathogenesis giving emphasis on those based on available literature are considered important for the malignant transformation of normal cells.
The JAK/STAT signaling pathway and cancer
As already mentioned the JAK/STAT signaling pathway transduces signals from cytokines and growth factors that act through a trans-membrane receptor families series [4]. Type I receptors include the erythropoietin (EPO) receptor and the granulocyte colony-stimulating factor (G-CSF) receptor. Type IIa receptor includes the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor, whereas the type IIb subfamily receptors includes those for leukemia inhibitory factor (LIF), and interleukin-6 (Il-6). Those receptors’ intracellular tails are constitutively associated with inactive kinases, known as Janus Kinases (JAKs). In some cases, signaling through STATs, which are molecular substrates of JAK kinases, can be activated by receptors having endogenous tyrosine kinase activity such as the PDGF-and EGF-receptor [91], although that activity sometimes also involves the cytoplasmic tyrosine kinase Src [92] (Figure 1).
Four members of the JAK family, JAK1-3 and TYK2 have been recognized in humans, and are able to generate homodimers and heterodimers, and also seven members of the STAT family have been revealed, STAT1-4, STAT5A, STAT5B and STAT6, all of which are typically located in the cytoplasm when inactive [93].
The JAK/STAT pathway activation, as in other signaling cascades, is controlled strictly by negative regulators acting at multiple levels [5,93]. (Figure 2). As mentioned its ingredients affect gene expression and can lead to chromatin epigenetic modifications [7]. Alterations in epigenetic modifications in cancer regulate various cellular responses, including cell proliferation, apoptosis, invasion, and senescence. Through DNA methylation, histone modification, chromatin remodeling, and noncoding RNA regulation, epigenetics play an essential role in tumorigenesis [94,95].
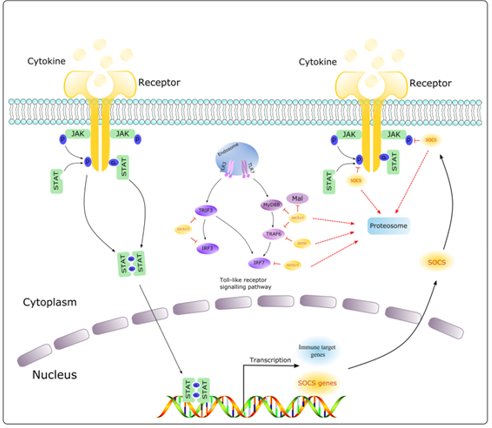
Figure 2. Pathways activating JAK/STAT signaling (Carcinogenesis, 2021)
STAT3 is highly expressed in tumor cells, due to the loss of precise negative regulation and persistent activation. It also induces the IL-6/JAKs [96], EGFR [97], Src [98] and many transcription factors accumulation and oncoproteins due to the inhibition or deletion of negative regulatory proteins, as well as over-stimulation of intracellular and extra-cellular factors. It also shows anti-apoptotic effect via directly up-regulating the Bcl-xl/ Mcl-1/ Bcl-w expression or induce indirectly the Hsp70/RegIIIβ apoptosis by the expression of heat shock protein Hsp70/ RegIIIβ- induction. Activated STAT3 is able to induce immediate anti-apoptosis by inducing the p100/p52 production, whereas it also suppresses p53-mediated apoptosis by binding to the promoter of p53 and inhibiting the p53 transcription. STAT3 promotes tumor cell proliferation by up-regulating the expression of the Cyclin B1/cdc-2/c-myc/cyclinD1/cjun/c-fos complex and down regulating the Cyclin B1 kinase inhibitor CDK1 expression in p21/p27 expressing cells, which accelerates the progression through the G1/S checkpoint [14,15]. STAT3 up-regulates a proliferation regulator, Cyclin D1. However, in HepG2 cells with high Cyclin D1 expression, STAT3 expression is suppressed, finding that has been confirmed in artificially inducible BC cells showing elevated Cyclin D1 expression. It is suggested that overactivated Cyclin D1 can negatively regulate the STAT3 activity [20].
JAK/STAT activation has been associated with diverse hematologic malignancies [9,10]. The recognition that the JAK2V617F mutation, was responsible for a CP phenotype in a mouse model and that STAT5A/ B is essential for its development in those mice gave evidence for a causative role for JAK2-STAT5 activation in MYNs. However, STAT3 role in those malignancies is not fully understood. STAT3 deletion in a mouse model increases MYN phenotype aspects and reduces survival suggesting that STAT3 may restrain malignant proliferation. This appears partly to be associated with secondary alterations in STAT1activation. In addition, it has been recorded that in MYNs, STAT3 may oppose malignant proliferation proposing that this may also occur in some solid tumors cases [99]. The accurate role of JAK/STAT signaling in tumorigenesis in MYNs and other cancer types has also been disputed by examination of rare cancer cases in families with germline mutations that are responsible for weak JAK activation [100,101]. To be more specific, the mutations are responsible for hereditary thrombocytosis, however hematopoiesis is a polyclonal process and individuals do not develop hematologic malignancies or solid tumors, suggesting that JAK/STAT activation itself is not able to lead to malignant disease [102]. In other hematological malignancies activating mutations affecting JAK/ STAT signaling have also been identified [103]. However, it remains to be determined, to what extent JAK/ STAT activation is the driving force for the mentioned diseases. An example concerns T-ALL in which in a significant proportion of patients JAK1 activating mutations were identified, and they were considered to promote the malignant cells survival, but JAK mutations were not considered to be the primary driving force of the disease [104]. JAK1 mutations may affect many hematological malignancies. To be more specific, the JAK2 V
617F, JAK1V658F JAK1 homolog, led to the kinase constitutive activation [10,21,22]. The JAK3M592F, homolog of JAK2V617F, does not lead to kinase constitutive activation as JAK3 is the only JAK family member that does not become constitutively activated by this mutation [21]. FERM domain mutations were rare as mutations with loss of function were associated with severe combined immunodeficiency (SCID), whereas gain-of-function mutations were associated with adult T-cell leukemia/lymphoma [105].
In almost 18. 4% of 38 T-ALL cases seven somatic heterozygous JAK1 missense mutations were found, but only 3. 4% of 88 B-ALL cases. Like mutations in other JAK family members, most of these unique JAK1 mutants concerned the pseudokinase domain and are therefore considered to interfere with kinase auto-inhibition and subsequent development of or progression to a lymphoid malignancy [106].
In primary mediastinal and Hodgkin lymphoma JAK2-mediated chromatin modification have been detected, both of which share common molecular and biological characteristics [30,31], as autocrine IL-13 production and chromosome 9p24 amplification both result to JAK2 activation.
The underlying mechanism of this oncogenic cooperation is H3Y41 phosphorylation by JAK2 along with H3K9 demethylation by JMJD2C, a coordinated chromatin modification process that releases HP1 suppression and consequently activates chromatin for gene transcription [107]. Epigenetic gene regulation by JAK1 has been detected in diffuse large B-cell lymphoma (DLB CL) [23]. A research on mouse embryonic stem cells detected that JAK1 was also present in the nucleus and could directly phosphorylate H3Y41 [108]. That finding was in agreement with the observation that JAK1 signaling in ABC DLBCL in which JAK1 is activated by the cytokines IL-6 and IL-10 [23]. In ABC DLBCL, JAK1 but not JAK2 is responsible for the phosphorylation of STAT3 and H3Y41 [109,110].
The JAK/STAT pathway activation in hematological malignancies is a result of gain-of-function mutations in JAKs kinases. Recent large-scale sequencing studies have revealed genetic alterations affecting JAKs in some cases of solid tumors. In a percentage of 9% of hepatitis B-associated hepatocellular carcinoma cases missense mutations in JAK1 have also been recorded, whereas a cell culture research has shown that those mutations increase JAK1 and STAT3 phosphorylation and can lead to cytokine-independent development. In gastric adenocarcinoma cases a comprehensive molecular process revealed frequent JAK2-containing chromosomal region amplification. Corresponding increases in JAK2 mRNA suggested that this may enhance JAK2 protein levels and pathway activity [111].
In many cancer cases in which JAK/STAT activation is a characteristic, the underlying mechanism of the inappropriate activation of the pathway it still remains unknown. Examining the cases in which a mechanism has been identified, it appears that malignant cells use a wide range of strategies to activate the pathway. The determination of the mechanisms that are responsible for JAK/STAT pathway activation do not indicate, however, whether the pathway activation contributes significantly to tumorigenesis in those cases [110]. G-CSF receptor increased expression has been recorded in cases of high-grade epithelial ovarian tumors, and cell culture experiments demonstrated that G-CSF contributes to JAK/STAT activation in that tumor types [112].
STATs activating mutations have been detected in cancer, although it is a rare finding. In PC cases, STAT5A/B locus amplification has been found and was associated with STAT5 increased expression and its nuclear localization in tumor samples. Those enhancements increased cell survival in cultures and promoted tumor growth in a xenograft model [12]. JAK/STAT increased activation could be a result of negative regulators decreased expression. In non-small cell lung cancer (NSCLC) samples, SOCS3 expression is lost due to promoter hypermethylation, an epigenetic alteration that decreases gene transcription. The effect of that event on pathway activation was examined using an NSCLC cell line, where SOCS3 expression restoration reduced constitutive STAT3 phosphorylation [113].
JAK/STAT pathway is also involved in glioblastoma progression, invasion, and migration. Glioma and other immune cells secrete IL-8, which promotes tumor invasion, migration, and mesenchymal transition by activating STAT1/hypoxia-inducible factor-1α(HIF-1α)/Snail pathway [114]. STAT5 signaling plays a role in invasion and proliferation in glioblastoma cases [19]. Moreover, glioblastoma patients had constitutively activated STAT3 and secreted IL-6 levels that were associated with tumor grade [115]. Protein Inhibitor of Activated Signal Transducer and Activator of Transcription 3 (PIAS3) protein levels have been found to be decreased in glioblastoma cases, probably because of increased protein degradation. In glioblastoma tissue samples, PIAS3 low levels were associated with increased pSTAT3 expression and proteins produced by STAT target genes [116].
STAT5 is responsible for protection against apoptosis by activating the transcription of Bcl-x to generate the anti-apoptotic protein Bcl-xL. Malignant cells undergo changes in energy metabolism, switching mitochondrial oxidative phosphorylation to glycolysis to generate ATP. STAT3 decreases the genes expression coding for mitochondrial proteins and increases the genes expression implicated in glycolysis, such as pyruvate dehydrogenase kinase 1. Those results depend on the HIF-1a transcription, that is induced by STAT3 [117], as it has a critical role in the adaptation of cancer cells to a hypoxic environment. STAT3 is required for the full transcriptional activation of HIF-1a-regulated genes in an hypoxic micro-environment [118]. Angiogenesis is essential for tumor growth, with a key role in which the VEGF factor participates. STAT3 binds to the VEGF promoter and induces its expression. In tumor allograft models, a constitutively active STAT3 expression results in increased VEGF expression and increased angiogenesis. A main characteristic in the interaction of malignant cells with the tumor microenvironment is their ability to suppress antitumor immune responses in case of hematologic malignancies [119]. Malignant cells in solid tumors, increase in number, adapt and change their microenvironment. Genes controlled by STATs play critical roles in both of those aspects of the malignant phenotype, although in many cases further investigation is required to define to what extent persistent JAK/STAT activation leads those phenotypic alterations and what contribution is made by other alterations in malignant cells. Cell cycle progression is facilitated by STAT3 by promoting the Cyclin-Dependent Kinases (CDKs) activation and increases the positive regulators transcription such as Cyclin D2 and down-regulates the CDK inhibitors transcription such as p21 [120].
The activation of STAT1 by interferons promotes immuno-surveillance and antitumor immunity, partly by up-regulating of MHC-I class-mediated antigen presentation by tumor cells. On the contrary, STAT3 and STAT5 signaling in immune cells seems to suppress antitumor immunity.
In mice, anti-tumor immune responses are increased by STAT3 knockdown from hematopoietic cells and by STAT3-blocking drugs [121]. The increased antitumor immunity revealed in those mice is due partly to a decrease in tumor-infiltrating regulatory T cells, whose their development depends on the STAT3/5 target gene, FOXP3 [122].
JAK/STAT activation has been associated with invasion and metastasis. This is partly mediated by the Epithelial-Mesenchymal Transition (EMT) program activation which is involved in embryonic development. TWIST1 is a transcription factor and an EMT induction key regulator.
TWIST1 overexpression induces EMT, a key process in the metastases formation of cancer, and also promotes cancer stem cells formation and facilitates tumorigenesis process. STAT3 is essential for the TWIST1 expression, whereas STAT3 activity abolition by siRNA knockdown, pharmacological inhibition, or by mutation of the STAT-binding site in the TWIST1 promoter, decreases its expression. STAT3 can activate the matrix-degrading enzymes transcription such as Matrix Metallo-Proteinase (MMP)-2 [123,124].
Although intracellular signaling pathways are often described as discrete pathways, are probably more accurately thought as networks consisting of multiple interactions between pathways. Physiological and functional affections have been reported for interactions between JAK/STAT signaling and an amount of other cellular signaling pathways is known to be implicated in tumorigenesis, including downstream signaling of the EGFR, and androgen receptor (AR) signaling.
The recognition of interactions between pathways has consequences for improving the effectiveness of targeted therapies by combining treatments that act on interacting pathways to overcome resistance. This has been shown in vitro, in melanoma models, where STAT3 is essential for full transcriptional activation of mutant B-RAF [125]. STAT3 phosphorylation suppression by STAT3siRNA knockdown or using a JAK2 inhibitor restores sensitivity to a B-RAF inhibitor in melanoma cell lines with acquired resistance to B-RAF inhibition [126].
The understanding of the way that pathway interactions lead to JAK-independent activation of STATs is also critical for treatment and may explain the JAK inhibitors failure in clinical trials in cases of solid tumors. Early evidence that JAK/STAT signaling is activated in solid tumors recorded from cancer cell lines. There is essential data showing STATs tyrosine phosphorylation and nuclear localization, indicative of their activation, in tumor tissue derived from a large number of patients with several tumor types [127]. For other tumor types, differences in the strategies used to quantify STAT phosphorylation, which vary across studies may are responsible for the seemingly conflicting associations between STAT phosphorylation and outcome [128]. In contrast, STAT1 activation, is generally associated with better outcomes in all tumor types. Although STAT activation has been found in a wide range of tumor types, in many cases, the mechanisms that cause its activation have not been clearly determined. Those studies present associations between JAK/STAT activation and outcomes, but do not define whether the observed JAK/STAT activation has a causal role in diseases. (Figure 3).
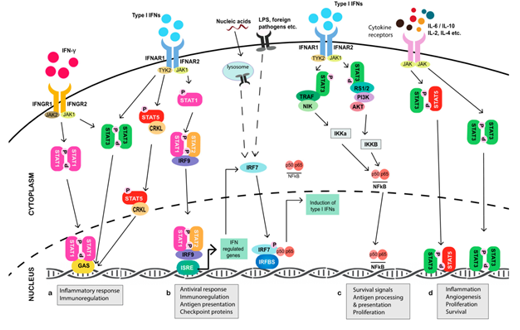
Figure 3. JAK/STAT signaling involvement in tumorigenesis mechanisms
JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression (Cancers – MDPI, 2019)
Notch signaling pathway and cancer
Notch family are trans-membrane proteins with dual functions, as act as membrane receptors and transcription factors. The Notch family in mammals, has four receptors, Notch1-4. Each receptor is composed in the endoplasmic reticulum as a precursor protein containing an intra-cellular, a trans-membrane, and an extracellular portion [32-34]. Five ligands for the Notch receptor have been identified so far, Delta 1, 3 and 4 (DLL1, 3 and 4) and Jagged1and 2 (JAG1 and 2), that are also trans-membrane proteins [34,129]. The pathway is activated when the ligand binds to the receptor of a neighboring cell. After activation, the receptors undergo a series of proteolytic cleavages by MMP TACE and the γ-secretase complex consisting of presenilin-1/2, nicastrin, Pen2, Aph1. TACE enzyme cleaves a portion of the receptor from its extracellular portion, that is internalized by the neighboring cell that has expressed the ligand [34]. (Figure 4 and 5).
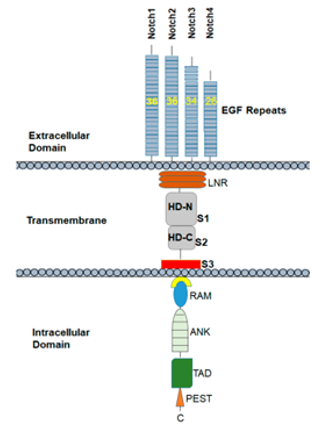
Figure 4. Notch receptor structure
(A Review on Notch Signaling and Colorectal Cancer (Cells – MDPI, 2020))
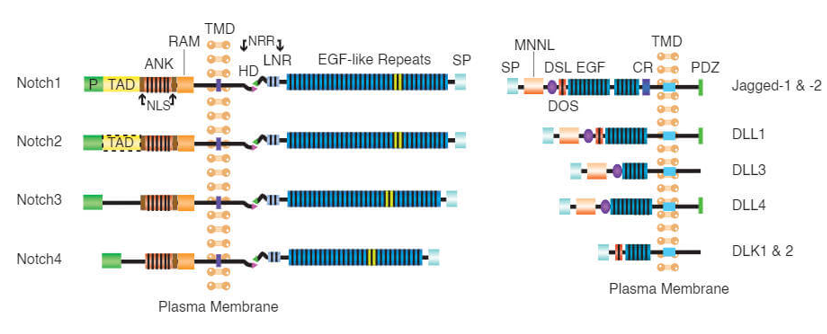
Figure 5. Notch receptor ligands (Creative Biomart)
The second part derived from the action of γ-secretase releases the intracellular part of Notch, NICD, into the cytoplasm that translocates to the nucleus where it activates the target genes transcription. After translocating to the nucleus, the NICD part binds to the transcriptional represssor cytoplasmic localization signal (CLS)and activates it. After the activation, it is converted into an active transcription factor and allows the initiation of target genes transcription. The main target genes of Notch consist of transcription factor families, such as Hes and Herp. The final stage of the NICD mechanism may involve a mechanism that is dependent on the CSL protein and involves the Hes/Herp proteins or is dependent on the CSL protein but does not involve the Hes/Herp proteins or is independent of the CSL protein. NICD may affect specific proteins using the above mechanisms. Moreover, it has been observed that NICD levels control the pathway. Since it controls several cell differentiation stages in different tissues, it is possible that it is implicated in different types of tumors development. Other target genes are Cyclin D1, NFκB, p21, GATA3, preTa, c-Myc, NRARP, and Deltex1 [129,130]. The Notch pathway may play a role in other pathways independent of the CSL factor, such as, it can promote the maturation of CD4+ and CD8+ T- cells of the thymus gland [130].
Dysregulated Notch signaling has been observed in many diseases and plays complex oncogenic or tumor-suppressive roles in different tumors depending on the tissue and cellular condition [47,49,131,132].
Notch pathway is closely related to ras, and its activation is likely a result of tumorigenesis mediated by the ras pathway. In ras-mutant fibroblasts, inhibition of the Notch pathway by γ-secretase inhibitors resulted in an 80% inhibition of tumor development. In the corresponding control cells, γ-secretase inhibition had no effect on tumor growth. In pancreatic cells, activation of the ras pathway by the transforming growth factor-alpha (TGF-α), led to Notch pathway activation and led also to a pro-tumor phenotype [133,134]. After its activation by the ras pathway, it triggers in turn the ras-raf-mek-MAPK segment that is a ras pathway branch. In small cell lung cancer (SCLC) cell cultures lacking ras mutations, where MAPK activity is generally low, and in SCLC cell cultures that have high MAPK activity and ras mutations, Notch1 activation led to high levels of pERK1and pERK2 [135]. Therefore, it can be considered that in some cases Notch activation may have a synergistic effect with ras. It is evident that the Notch interaction and the ras pathway may be an important field of research in NSCLC cases where ras mutations exist, enhanced EGFR activity, and activated ligand activity of the ras pathway [136].
Other signaling pathways that Notch pathway interacts are NFκB, Wnt, Akt, mTOR, Sonic Hedgehog, PDGF, estrogen and androgen receptor [137] (Figure 6).
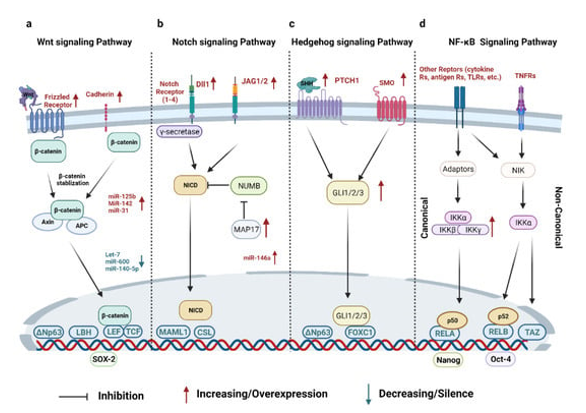
Figure 6. Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic
As mentioned, Notch pathway plays a crucial role in survival, cell proliferation, differentiation, and apoptosis, processes that affect the development and function of many organs. Notch pathway dysfunction prevents differentiation leading undifferentiated cells to malignant transformation. Scientific observations suggested that the Notch pathway alterations are associated with many cancers, such as breast, lung, ovarian cancer and leukemia. In addition, its receptors and ligands may be prognostic or diagnostic markers for cancer [40].
Patients with tumors that express high levels of Jagged1 or Notch1 have a shorter survival rate than those that do not express high levels of those genes. High expression of Jagged1 in prostate cancer (PC) is significantly associated with tumor recurrence and metastasis suggesting that Jagged1 may be a useful marker in distinguishing between aggressive and non-aggressive tumors in (PC) cases. High co-expression levels of Jagged1 and Notch1 were observed in BC and found to be associated with short survival rate. Therefore, the expression of Notch1 and Jagged1may be markers for patient survival. Cervical cancer patients with Notch3 expression had a significantly shorter survival rate than those without Notch3 expression. Therefore, Notch3 could be a prognostic marker in this type of cancer [137,138]. Jagged1 has been associated with HCC. Recently was recorded that Jagged1 expression was observed in 83 of 145 HCC cases, with significantly increased levels detected in tumor tissues than those in adjacent non-tumor tissues [139]. In another study of 112 HCC specimens, membrane localized Jagged1 (JAG1Mem) expression was observed in 39 (34. 82 %) specimens and cytoplasmic Jagged1 (JAG1Cyto) expression was found in 73 (65. 18 %) specimens. Furthermore, Jagged1Mem interacts with Notch1Mem and leads to Notch1 signaling activation, which up-regulates osteopontin (OPN), an essential extra-hepatic HCC metastasis driver [140].
Concerning the association between Jagged1 in gastric carcinoma, immune-histochemical (IHC) staining showed that 72 of 96 (75 %) gastric carcinoma (GC) cases expressed Jagged1 in cancer tissues, and that this expression was associated with advanced cancer [141]. Analyses in gene and protein expression also detected higher Jagged1 expression in GC tissues and cells than in adjacent tissues and normal gastric epithelial cells [142].
Notch signaling was considered oncogenic in glioma cases, in which it maintains brain cancer stem cells [52]. In vitro and in vivo [143] was found that Notch signaling inhibition with a γ-secretase inhibitor in glioma or Notch ligands in human brain micro-vascular endothelial cells knockdown restricted tumor development [144]. IHC staining in several studies showed high Jagged1 expression in glioma tissues distributed in the membrane, and tumor cells cytoplasm and vascular endothelial cells, whereas no significant Jagged1staining was found in adjacent normal brain tissues [145-147]. An EGFR variant, EGFRvIII, leads aberrant MAPK signaling such as MEK/ERK, and its expression levels associate positively with Jagged1 expression levels in glioblastoma cases. Jagged1/Notch signaling stimulated by the EGFRvIII/MEK/ERK axis maintains the stem cell-like property of glioma-initiating cells (GICs) [148]. In addition, Notch1 signaling facilitates the glioma-initiating cells invasion and development by modulating the CXCL12/ CX CR4 chemokine pathway [149], whereas inactivation of Rbpj, Notch1, or Notch2 receptors accelerates tumor development in a mouse model [150]. That subtype-specific effect of Notch in glioma cases could be attributed to cooperation with p53. In conclusion, Notch signaling acts either as an oncogenic promoter or a tumor suppressor in different glioma subtypes, and the underlying mechanisms need further research [151].
Jagged1 overexpression is widely reported in BC and is associated with a very poor prognosis in BC cases, and is also associated with high pathological grade, advanced tumor stage, lymph node metastasis, co-expression with the Ki-67 tumor marker, and LDH1 [152-154]. However, Jagged1 roles are different in different BC molecular subtypes. The Jagged1/Notch-induced EMT process and metastasis in cases of advanced BC are promoted by non-tumor cells in the tumor microenvironment, including tumor-associated macrophages (TAMs) [153,155], fibroblasts [156], tumor endothelial cells [157], and bone stromal cells, pro-osteoclasts and osteoblasts [157-159]. BC tissues were the first epithelial tumors examined regarding Notch signaling [160-162]. An important observation was that the Notch1 and Jagged1up-regulation, known as non-mutated Notch signaling-related proteins were associated with poor prognosis in BC cases [163]. Mutations in Notch1 and Notch4, on mouse models, mediated by mouse mammary tumor viruses were able to promote epithelial mammary tumorigenesis [164]. Notch activation is associated with cytotoxic chemotherapy resistance in HER2- expressing BC cells [165] and could be attributed to a lack of NUMB expression [166] and several reasons such as the fact that Notch signaling maintains the BC cells stemness and promotes initiation [167,168], and it forms features of the BC microenvironment, especially TAMs, that is associated with the innate immune phenotype [169].
Approximately 23% of OC patients had Notch signaling alterations according to the Cancer Genome Atlas Research Network [54], whereas Notch1 and Notch3 promote OC development [54-56]. Moreover, Notch3 overexpression was associated with cell hyperproliferation and apoptosis inhibition, tumor metastasis and recurrence [170,171]. Notch3 was positively associated with Jagged1and Jagged2 expression in OC, and the Notch3 carcinogenic function is potentially mediated by Jagged1-Notch3 activation [172]. Also, Notch signaling promotes angiogenesis in OC mediated by VEGFR2 negative feedback [173] through the VEGFR2 promoter methylation.
In a study of 89 OC cases qPCR analysis and IHC staining detected that Jagged1was highly expressed in high-grade OC tissues compared with low-grade OC tissues [172]. In another study, qPCR and Western blot analysis suggested that Jagged1 expression was up-regulated in most OC cell lines compared with normal ovarian cell lines [174].
Active Notch signaling plays an oncogenic role in CRC and furthermore, Jagged1 and DLL4 may control Notch activation at different tumor stages [42]. In vitro and in vivo findings suggested that Jagged1 contributes to the CRC cells proliferation and invasion and tumor growth [43]. Notch oncogenic effects regarding hematological malignancies were focused on chromosome t (7;9) translocation of the Notch1 gene in T-ALL, whereas more than of 50% of T-ALL cases had Notch1 somatic activating mutations [50]. In murine models transplanted hematopoietic progenitor cells with constitutive activation of Notch1 signaling led to T-ALL development [175]. Similar data suggested that Notch1 may induce the myc expression by regulating its enhancer N-Me and play a critical role in the T-ALL initiation and maintenance [176]. The interaction of Notch1 and Pten pathways promote anabolic processes in T-ALL cases, as a synergistic effect [177]. Moreover, Notch1 can directly regulate specific lncRNAs expression, such as LU NAR1, which is essential for the T-ALL cells [178] malignant proliferation, whereas Notch signaling regulates the T-ALL cell cycle progression via G1 phase proteins CyclinD3 expression, CDK4, and CDK6 [179]. Recently it was found that, Notch3 activating mutations independent of Notch1 ones had also been detected in several T-ALL cases [180]. In other hematological malignancies activating mutations in Notch have also been observed. In a murine model with B cell chronic lymphocytic leukemia (BCLL), Notch signaling dysfunction reduced morbidity, whereas Notch signaling activation increased the survival and apoptosis resistance of B-CLL cells [181].
Notch also was responsible for tumor growth through the FBXW7-NOTCH- CCL2/CSF1 axis [182] in diffuse large B-cell lymphoma (DLBCL) cases. Despite the fact that Notch has an oncogenic role in most hematological malignancies, it inhibits AML growth and survival and Notch1-4 consistent activation led to AML growth arrest and caspase-dependent apoptosis [183].
The Notch1 -3 tumorigenesis effects might implicate downstream genes activating mutations regulating the tumor-initiating cell phenotype. In a mouse model [63] was observed that Notch1 and myc co-activation increased the NICD1-induced adenoma formation frequency and facilitated tumor progression and metastases. Moreover, Notch3 was an essential gene in KRAS-mediated LA that activates PKCi-ELF3-NOTCH3 signaling for regulation the asymmetric cell division in tumor initiation and maintenance processes [184] whereas Notch1 activation in KRAS-induced LA suppressed p53-mediated apoptosis [61]. However, a recent report showed that Notch mutations had opposite effects in LA and squamous cell carcinoma (SCC) [185], therefore the specific role of Notch in LA cases needs further research.
Notch signaling is essential for the development and homeostasis of normal intestinal epithelial cells, as regulates the colonic goblet cells and stem cells [35] differentiation [36], under normal circumstances. In CRC tissues, notable Notch ligands (DLL1, DLL3, DLL4, JAG1, and JAG2) up-regulation and Notch receptor (Notch1) abnormal activation have been found [186], which led to metastasis and poor prognosis of CRC [187]. It has been suggested that Notch activation is a trigger of colon cancer development, as Notch inhibition by miR-34a and Numb suppressed the colon cancer stem cells [188] proliferation and differentiation. Aberrant Notch signaling facilitates the CRC cells invasion and metastasis probably through the NOTCHDAB1-ABL-TRIO pathway, EMT and TGF-β-dependent neutrophil effects [189]. In addition, Notch signaling also facilitates CRC invasion by inducing ABL tyrosine kinase activation and the RHOGEF protein TRIO [190] phosphorylation, whereas active Notch signaling promotes the metastasis by reforming the tumor microenvironment and regulating EMT-associated transcription factors such as SNAIL and SLUG [187,191,192] and induces EMT in colon cancer cells with TP53 deletion [190,193].
Notch signaling is a pathogenic factor in non-alcoholic steatohepatitis (NASH), however its role in HCC pathogenesis still remains unclear [194]. Activated Notch signaling has been observed in approximately 30% of human HCC samples which in mice led to the liver tumor formation [195]. Notch activation is also associated with the insulin-like growth factor 2 (IGF-2) activation that contributes to hepatocellular carcinogenesis [196], whereas it promotes EMT progression and metastasis in HCC [197]. However, Notch activation slows HCC development and can predict its prognosis [198]. Myb signaling up-regulation through Notch mutation and amplification might be a potential ACC driving mechanism [199], whereas activated Notch1 also generates CD133(+) ACC cells, that are known as cancer stem-like cells in ACC cases.
The Notch ligands and receptors overexpression, in clear cell renal cell carcinoma (CCRCC), has been observed in tumor tissues. Notch1 activation is responsible for dysplastic tubular epithelial cells hyperproliferation, whereas treatment implicating a γ-secretase inhibitor resulted in CCRCC cell inhibition in vitro and in vivo [69].
Notch may be implicated in many cancer types as a pro-tumor effector, however it can also act as a tumor suppressor in others, such as in SCC and neuroendocrine tumors [76] (Tables 1 and 2). Antitumor mechanisms concern regulation of transcription factors with malignant properties, suppressive genes downstream activation, cell cycle inhibition, etc., in general the traditional role of Notch as an oncogene has been challenged [151].
Notch is considered to act as a suppressor in neuroendocrine tumors (NETs), such as tumors derived from the thyroid, intestine-pancreas and the respiratory system neuroendocrine cells [200].
SCLC is the most frequent lung NET type, as 25% of human SCLC cases showed Notch target genes inactivation in one comprehensive genomic profiling analysis [201]. A recent study found that the Notch signaling down-regulation was essential for the initial cell state switch of LA cells [202], suggesting that Notch has a tumor-suppressive role in SCLC cases. Moreover, high DLL3 expression was frequently found in lung carcinoid tumors and SCLC cases [202-204], which down-regulates Notch signaling via cis-inhibition. Notch1 or Notch2 activation in an SCLC mouse model, reduced the synaptophysin and Ascl1 expression, inhibiting the cell cycle process [205,206]. Similarly, in human medullary thyroid cancer (MTC) tumor samples, Notch1 protein was undetectable, whereas the NICD1 expression of inhibited MTC cell proliferation [207]. In a gastro-intestinal-pancreatic NET tumor specimens analysis, reduced Notch expression and mutated components were observed [208,209]. Some researches suggested that such an anti-tumorigenesis effect could be mediated by the NOTCH-ASCL1-RB-P53 tumor suppression pathway [210,211], whereas others proposed that activated Notch could inhibit cell development via cell cycle arrest associated with p21 up-regulated [207,212]. Notch could also mark and initiate deprogramming in rare pulmonary NET cells that serve as stem cells in SCLC. Considering the suppressor effect of Notch in NETs, drugs targeting DLL3 have been tested in SCLC, with promising results witnessed in preclinical trials [213].
Data concerning the mutated Notch in SCCs showed that Notch function relies on aspects such as, Notch function can be affected by the p53 pathway and the intrinsic transcription-repressive protein RBP-Jκ [82]. The underlying regulatory mechanism is unclear, although some reports suggested that Notch signaling maintains the CD133 phenotype in SCCs stem cell [214]. Moreover, decreased Notch1 expression also dysregulates cell cycle-associated genes in SCCs such as LSC [185].
Growing data demonstrated frequent NOTCH2 mutations in splenic marginal zone lymphoma (SMZL) focusing its critical role in the pathogenesis of this disease [215].
The Notch pathway regulates pancreatic cell differentiation [216] and is involved in the development and progression of PDAC [217-220]. Although previous articles have recorded the Notch signaling components activation in PDAC [87,221-224], the association between increased Notch expression and tumorigenesis in PDAC is controversial as contradictory findings have been reported by different studies. Mazur et al. [218] revealed that Notch signaling has a tumor promoting effect, whereas Hanlon et al. [87] demonstrated that it had an inhibitory tumor effect [218,224].
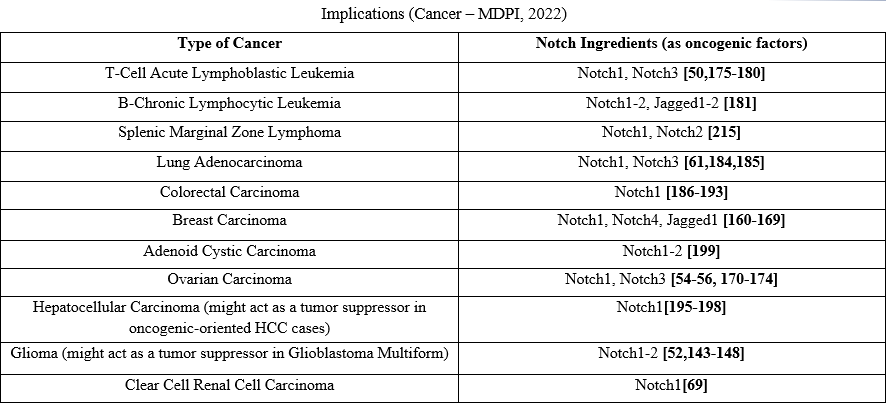
Table 1. Notch signaling pathways in Carcinogenesis acting as an oncogenic factor
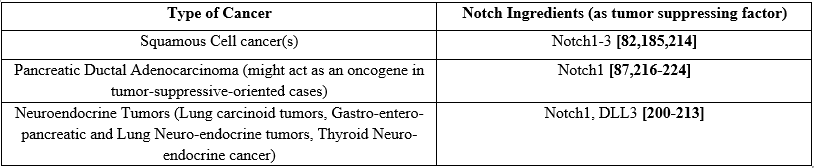
Table 2. Notch signaling pathways in Carcinogenesis acting as tumor suppressing factor
EMT is responsible for increased stemness in tumors and several cancer models have revealed that EMT plays an essential role for initiating metastasis trigger [225-227]. Jagged-1, a Notch1 ligand is implicated in metastasis in PC, BC, and CRC and the Notch1 signaling activation contributes to cancer cell stemness and invasion in the mentioned cancer cases [159,228]. Wieland et al. [229] successfully showed that the maintained Notch1 activity in epithelial cells is responsible for a senescence-like phenotype development, which eventually facilitates tumor cells trans-mitgration within the primary tumor and homing at distant locations-metastasis. In a similar way, Notch1over-expression in immortalized endothelial cells induces Snail expression, reduces E-cadherin expression that finally leads to the contact inhibition loss and the EMT acquisition followed by oncogenic transformation. Jagged1 induces EMT, which, in turn, regulates Slug, that is responsible for EMT. In human BC cells it has been observed that Notch1 inhibition is able to invert the Jagged1-induced EMT process [227]. Notch signaling activation induced by Jagged1 led to the inhibition of E-cadherin expression that damages cell-cell adhesion and a simultaneous increase in N-cadherin, nuclear localization of β-catenin and vimentin, that eventually led to an invasive and mesenchymal phenotype [230].
In BC, the rate of N1ICD-positive versus negative tumor epithelial cells (80% ± 10. 3% versus 50% ± 12. 5%) was associated with patients who had positive sentinel lymph nodes. In melanoma, N1ICD expression was significantly associated with higher rates of metastasis (stage IV tumors) and shorter progression-free survival compared to patients with low N1ICD expression [229]. Those studies suggested that Notch1 activation plays a major role in EMT and, when activated in the vasculature, presents a major risk factor for metastasis.
Conclusions
Previous reports have shown that a large amount of solid tumors exhibit JAK/ STAT signaling pathway activation. The central position of JAK/STAT signaling in a signaling pathways network whose deregulation contributes to cancer suggests that targeted inhibition of JAK/ STAT signaling could be utilized therapeutically to treat solid tumor cases. Notch signaling alterations play a very important role in several aspects of carcinogenesis. The investigation of the Notch core and target gene expression patterns in diverse cancer types resulted in the deep exploration of biological differences. Specific differences between described cancer types can be found in the comparison of neoplastic and normal tissues, especially for the Notch ligands and its receptors expression. The absence or low frequency of mutations and copy number variation in core Notch members may display that they have insignificant impact for Notch signaling differentiation between different cancers. Notch expression activation depends on tissue conditions, inhibiting trans formation in some tissues and promoting tumorigenesis in others. Furthermore, the variety in consecutive stages of Notch signaling affected the target genes expression profile, as reflected in the various genes activity involved in basic cellular functions such as proliferation, adhesion, EMT or apoptosis. All those observations provide new pathways for targeted therapy development. Depending on the tissue therapeutic intervention signs may contain igands, receptors, transcription factors, and regulators.
Conflict of interest and source of funding statement:
The authors declare that they have no conflict of interest
Dr. Nikolaos A. Chrysanthakopoulos
DDSc, Implant Surgeon (Cert. Att), Oncologist (MSc Oncol), PhD in Oncology (cand), RN.
References
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, et al. (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 439: 353-357. [PubMed.]
- Hanahan D, Weinberg RA. (2011) Hallmarks of cancer: the next generation. Cell. 144: 646-674. [Ref.]
- Witsch E, Sela M, Yarden Y. (2010) Roles for growth factors in cancer progression. Physiol (Bethesda). 25: 85-101. [Ref.]
- Aaronson DS, Horvath CM. (2002) A road map for those who don't know JAK-STAT. Science. 296 (5573): 1653-1655. [PubMed.]
- Morris R, Kershaw NJ, Babon JJ. (2018) The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 27(12): 1984-2009. [PubMed.]
- Kiu H, Nicholson SA. (2012) Biology and significance of the JAK/STAT signaling pathways. Growth Fact. 30: 88-106. [Ref.]
- Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, et al. (2009) JAK2phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 461: 819-822. [PubMed.]
- Girodon F, Steinkamp MP, Cleyrat C, Hermouet S, Wilson BS. (2011) Confocal imaging studies cast doubt on nuclear localization of JAK2V617F. Blood. 118: 2633-2634. [Ref.]
- Vainchenker W, Constantinescu SN. (2013) JAK/STAT signaling in hematological malignancies. Oncogene. 32: 2601-2613. [PubMed.]
- Bellanger D, Jacquemin V, Chopin M, Pierron G, Bernard OA, et al. (2014) Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 28(2): 417-419. [PubMed.]
- Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, et al. (2012) Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia. N Engl J Med. 366: 1905-1913. [PubMed.]
- Haddad BR, Gu L, Mirtti T, Dagvadorj A, Vogiatzi P, et al. (2013) STAT5A/B gene locusundergoes amplification during human prostate cancer progression. Am J Pathol. l182: 2264-2275. [Ref.]
- Siveen KS, Sikka S, Surana R, Dai X, Zhang J, et al. (2014) Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 1845: 136-154. [PubMed.]
- Hu B, Zhang K, Li S, Li H, Yan Z, et al. (2016) HIC1 attenuates invasion and metastasis by inhibiting the IL-6/STAT3 signaling pathway in human pancreatic cancer. Cancer Lett. 376: 387-398. [PubMed.]
- Nakamura H, Taguchi A, Kawana K, Kawata A, Yoshida M, et al. (2016) STAT3 acivity regulates sensitivity to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in cervical cancer cells. Int J Oncol. 49: 2155-2162. [PubMed.]
- Aleskandarany MA, Agarwal D, Negm OH, Ball G, Elmouna A, et al. (2016) The progno-stic significance of STAT3 in invasive breast cancer: analysis of protein and mRNA expressions in large cohorts. Breast Cancer Res Treat. 156: 9-20. [PubMed.]
- Wu WY, Li J, Wu ZS, Zhang CL, Meng XL. (2011) STAT3 activation in monocytes accelerates liver cancer progression. BMC Cancer. 11: 506. [Ref.]
- Lesinski GB. (2013) The potential for targeting the STAT3 pathway as a novel therapy for melanoma. Future Oncol. 9: 925-927. [PubMed.]
- Cao S, Wang C, Zheng Q, Qiao Y, Xu K, et al. (2011) STAT5 regulates glioma cell invasion by pathways dependent and independent of STAT5 DNA binding. Neurosci Lett. 487(2): 228-233. [PubMed.]
- Sinibaldi D, Wharton W, Turkson J, Bowman T, Pledger WJ, et al. (2000) Induction of p21 WAF1/CIP1 and cyclin D1expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene. 19: 5419-5427. [PubMed.]
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, et al. (2005) A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera. Nature. 434(7037): 1144-1148. [PubMed.]
- Jeong EG, Kim MS, Nam HK, Min CK, Lee S, et al. (2008) Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 14(12): 3716-3721. [PubMed.]
- Rui L, Drennan AC, Ceribelli M, Zhu F, Wright GW, et al. (2016) Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 113(46): E7260-E7267. [PubMed.]
- Bains T, Heinrich MC, Loriaux MM, Beadling C, Nelson D, et al. (2012) Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia. 26(9): 2144-2146. [PubMed.]
- Bouchekioua A, Scourzic L, de Wever O, Zhang Y, Cervera P, et al. (2014) JAK3 deregulation by activating 1. Bouchekioua A, Scourzic L, de Wever O, Zhang Y, Cervera P, et al. (2014) JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 28(2): 338-348. [PubMed.]
- Koo GC, Tan SY, Tang T, Poon SL, Allen GE, et al. (2012) Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2(7): 591-597. [PubMed.]
- Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, et al. (2013) Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 45(8): 937-941. [PubMed.]
- Lopez C, Bergmann AK, Paul U, Murga Penas EM, Nagel I, et al. (2016) Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol. 173(2): 265-273. [PubMed.]
- Wintering A, Dvorak CC, Stieglitz E, Loh ML. (2021) Juvenile myelomonocytic leukemia in the molecular era: a clinician’s guide to diagnosis, risk stratification, and treatment. Blood Adv. 5: 4783-4793. [PubMed.]
- Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, et al. (2003) Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 198(6): 851-862. [PubMed.]
- Savage KJ, Monti S, Kutok JL, Cattoretti G, Neuberg D, et al. (2003) The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 102(12): 3871-3879. [PubMed.]
- Miele L. (2006) Notch signaling. Clin Cancer Res. 12: 1074-1079. [PubMed.]
- Urata Y, Takeuchi H. (2020) Effects of Notch glycosylation on health and diseases. Dev Growth Differ. 62: 35-48. [Ref.]
- Wang MM. (2011) Notch signaling and Notch signaling modifiers. Int J Biochem Cell Biol. 43: 1550-1562. [PubMed.]
- Qiao L, Wong BC. (2009) Role of Notch signaling in colorectal cancer. Carcinogenesis. 30: 1979-1986. [PubMed.]
- Tyagi A, Sharma AK, Damodaran C. (2020) A review on Notch signaling and colorectal cancer. Cells. 9: 1549. [PubMed.]
- Wang Z, Li Y, Banerjee S, Sarkar FH. (2009) Emerging role of Notch in stem cells and cancer. Cancer Lett. 279: 8-12. [Ref.]
- Ferrando AA. (2009) The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program. 353-361. [PubMed.]
- Aster JC, Pear WS, Blacklow C. (2008) Notch Signaling in Leukemia. Annu Rev Pathol. 3: 587-613. [PubMed.]
- Perez-Fidalgo JA, Ortega B, Simon S, Samartzis EP, Boussios S. (2020) NOTCH signaling in ovarian cancer angiogenesis. Ann Transl Med. 8(24): 1705. [Ref.]
- Zhou B, Lin W, Long Y, Yang Y, Zhang H, et al. (2022) Notch signaling pathway: architeture, disease, and therapeutics. Sign Transduc Targ Ther. 7: 95. [Ref.]
- López-Arribillaga E, Rodilla V, Colomer C, Vert A, Shelton A, et al. (2018) Manic Fringe deficiency imposes Jagged1 addiction to intestinal tumor cells. Nat Commun. 9: 2992. [Ref.]
- Dai Y, Wilson G, Huang B, Peng M, Teng G, et al. (2014) Silencing of Jagged1 inhibits cell growth and invasion in colorectal cancer. Cell Death Dis. 5: e1170. [Ref.]
- Aster JC, Pear WS, Blacklow SC. (2017) The Varied Roles of Notch in Cancer. Annu Rev Pathol Mech Dis. 12: 245-275. [PubMed.]
- Sorrentino C, Cuneo A, Roti G. (2019) Therapeutic targeting of notch signaling pathway in hematological malignancies. Mediterr J Hematol Infect Dis. 11(1): e2019037. [Ref.]
- Arruga F, Vaisitti T, Deaglio S. (2018) The NOTCH pathway and its mutations in mature B cell malignancies. Front Oncol. 8: 550. [Ref.]
- Goriki A, Seiler R, Wyatt AW, Contrerassanz A, Bhat A, et al. (2018) Unravelling disparate roles of NOTCH in bladder cancer. Nat Rev Urol. 15(6): 345-357. [PubMed.]
- Zhang M, Biswas S, Qin X, Gong W, Deng W, et al. (2016) Does Notch play a tumor suppressor role across diverse squamous cell carcinomas? Cancer Med. 5: 2048-2060. [Ref.]
- Huang T, Zhou Y, Cheng AS, Yu J, To KF, et al. (2016) NOTCH receptors in gastric and other gastrointestinal cancers: oncogenes or tumor suppressors? Mol Cancer. 15(1): 80. [Ref.]
- Weng AP, Ferrando AA, Lee W, Morris 4th JP, Silverman LB, et al. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 306: 269-271. [PubMed.]
- Bonfiglio F, Bruscaggin A, Guidetti F, di Bergamo LT, Faderl M, et al. (2022) Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood. 139: 732-747. [PubMed.]
- Hu YY, Zheng MH, Cheng G, Li L, Liang L, et al. (2011) Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer. 11: 82. [Ref.]
- Natarajan S, LiY, Miller EE, Shih DJ, Taylor MD, et al. (2013) Notch1-induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res. 73: 5381-5390. [PubMed.]
- The Cancer Genome Atlas Research Network. (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609-615. [PubMed.]
- Park JT, Li M, Nakayama K, Mao T-L, Davidson B, et al. (2006) Notch3 gene amplification in ovarian cancer. Cancer Res. 66: 6312-6318. [PubMed.]
- Hopfer O, Zwahlen D, Fey MF, Aebi S. (2005) The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer. 93: 709-718. [Ref.]
- Zhu H, Zhou X, Redfield S, Lewin J, Miele L. (2013) Elevated Jagged-1 and Notch-1expression in high grade and metastatic prostate cancers. Am J Transl Res. 5: 368-378. [Ref.]
- Yu Y, Zhang Y, Guan W, Huang T, Kang J, et al. (2014) Androgen receptor promotes the oncogenic function of overexpressed Jagged1 in prostate cancer by enhancing cyclin B1 expression via Akt phosphorylation. Mol Cancer Res. 12: 830-842. [PubMed.]
- Yuan X, Wu H, Xu Han, Han N, Chu Q, et al. (2015) Meta-analysis reveals the correlation of Notch signaling with non-small cell lung cancer progression and prognosis. Sci Rep. 5: 10338. [Ref.]
- Liu L, Tao T, Liu S, Yang X, Chen X, et al. (2021) An RFC4/Notch1 signaling feedback loop promotes NSCLC metastasis and stemness. Nat Commun. 12: 2693. [Ref.]
- Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, et al. (2013) Notch1 is required or Kras-induced flung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 73: 5974-5984. [Ref.]
- Xu X, Huang L, Futtner C, Schwab B, Rampersad RR, et al. (2014) The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes Dev. 28: 1929-1939. [Ref.]
- Allen TD, Rodriguez EM, Jones KD, Bishop JM. (2011) Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 71: 6010-6018. [Ref.]
- Westhoff B, Colaluca IN, D’ Ario G, Donzelli M, Tosoni D, et al. (2009) Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci USA. 106: 22293-22298. [PubMed.]
- Ho AS, Kannan K, Roy DM, Morris LGT, Ganly I, et al. (2013) The mutational landscape of adenoid cystic carcinoma. Nat Genet. 45: 791-798. [PubMed.]
- Drier Y, Cotton MJ, Williamson KE, Gillespie SM, Ryan RJH, et al. (2016) An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 48: 265-272. [PubMed.]
- Karpinets TV, Mitani Y, Liu B, Zhang J, Pytynia KB, et al. (2021) Whole-genome sequencing of common salivary gland carcinomas: subtype-restricted and shared genetic alterations. Clin Cancer Res. 27: 3960-3969. [PubMed.]
- Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, et al. (2018) A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. 29: 1561-1568. [PubMed.]
- Bhagat TD, Zou Y, Huang S, Park J, Palmer MB, et al. (2017) Notch pathway is activated via genetic and epigenetic alterations and is a therapeutic target in clear cell renal cancer. J Biol Chem. 292: 837-846. [PubMed.]
- South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, et al. (2014) NOTCH1mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 134: 2630-2638. [PubMed.]
- Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, et al. (2011) Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA. 108: 17761-17766. [PubMed.]
- Huang T, Zhou Y, Cheng A, Yu J, To K-F, et al. (2016) NOTCH receptors in gastric and other gastrointestinal cancers: Oncogenes or tumor suppressors? Mol Cancer. 15: 80. [Ref.]
- Revandkar A, Perciato ML, Toso A, Alajati A, Chen J, et al. (2016) Inhibition of Notch pathway arrests PTEN-deficient advanced prostate cancer by triggering p27-driven cellular senescence. Nat Commun. 7: 13719. [PubMed.]
- Hristova NR, Tagscherer KE, Fassl A, Kopitz J, Roth W. (2013) Notch1-dependent regulation of p27 determines cell fate in colorectal cancer. Int J Oncol. 43: 1967-1975. [PubMed.]
- Bol G, Raman V, Van Der Groep P, Vermeulen JF, Patel AH, et al. (2013) Expression of the RNA Helicase DDX3 and the Hypoxia Response in Breast Cancer. PLoS One. 8: e63548. [Ref.]
- Nowell CS, Radtke F. (2017) Notch as a tumour suppressor. Nat Rev Cancer. 17: 145-159. [PubMed.]
- Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, et al. (2014) A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med. 20: 1199-1205. [PubMed.]
- Gao YB, Chen Z-L, Li J-G, Hu X-D, Shi X-J, et al. (2014) Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 46: 1097-1102. [PubMed.]
- Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, et al. (2012) Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2: 899-905. [PubMed.]
- Khelil M, Griffin H, Bleeker MCG, Steenbergen RDM, Zheng K, et al. (2021) Delta-like ligand-Notch1 signaling is selectively modulated by HPV16 E6 to promote squamous cell proliferation and correlates with cervical cancer prognosis. Cancer Res. 81: 1909-1921. [PubMed.]
- Natsuizaka M, Whelan KA, Kagawa S, Tanaka K, Giroux V, et al. (2017) Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat Commun. 8: 1758. [PubMed.]
- Al Labban D, Jo S-H, Ostano P, Saglietti C, Bongiovanni M, et al. (2018) Notch-effector CSL promotes squamous cell carcinoma by repressing histone demethylase KDM6B. J Clin Investig. 128: 2581-2599. [PubMed.]
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 333: 1154-1157. [PubMed.]
- Fukusumi T, Califano JA. (2018) The NOTCH pathway in head and neck squamous cell carcinoma. J Dent Res. 97: 645-653. [PubMed.]
- Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, et al. (2016) Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 531: 47-52. [PubMed.]
- Avila JL, Kissil JL. (2013) Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 19: 320-327. [Ref.]
- Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, et al. (2010) Notch1 functions as a tumor suppressor in a model of K-ras induced pancreatic ductal adenocarcinoma. Cancer Res. 70: 4280-4286. [PubMed.]
- Plentz R, Park J-S, Rhim AD, Abravanel D, Hezel AF, et al. (2009) Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 136: 1741-1749. e1746. [PubMed.]
- Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, et al. (2011) Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J Clin Invest. 121: 4685-4699. [PubMed.]
- Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, et al. (2012) Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med 209: 437-444. [Ref.]
- Silvennoinen O, Schindler C, Schlessinger J, Levy DE. (1993) Ras-independent growth factor signaling by transcription factor tyrosine phosphorylation. Science. 261: 1736-1739. [PubMed.]
- Niu G, Bowman T, Huang M, Shivers S, Reintgen D, et al. (2002) Roles of activated Srcand Stat3 signaling in melanoma tumor cell growth. Oncogene. 21: 7001-7010. [PubMed.]
- Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. (2015) The role of JAK/STAT signaling in the pathogenesis, prognosis and treatment of solid tumours. Brit J Cancer. 113: 365-371. [PubMed.]
- Hu X, Dutta P, Tsurumi A, Li J, Wang J, et al. (2013) Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc Natl Acad Sci USA. 110: 10213-10218. [Ref.]
- Bure IV, Nemtsova MV, Kuznetsova EB. (2022) Histone Modifications and Non-Coding RNAs: Mutual Epigenetic Regulation and Role in Pathogenesis. Int J Mol Sci. 23(10): 5801. [Ref.]
- Zhao G, Zhu G, Huang Y, Zheng W, Hua J, et al. (2016) IL-6 mediates the signal pathway of JAK-STAT3-VEGF-C promoting growth, invasion and lymph angiogenesis in gastric cancercer. Oncol Rep. 35: 1787-1795. [PubMed.]
- Andersen P, Pedersen MW, Woetmann A, Villingshoj M, Stockhausen MT, et al. (2008) EGFR induces expression of IRF-1 via STAT1 and STAT3 activation leading to growth arrest of human cancer cells. Int J Can. 122: 342-349. [PubMed.]
- Harada D, Takigawa N, Kiura K. (2014) The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers (Basel). 6: 708-722. [Ref.]
- Grisouard J, Shimizu T, Duek A, Kubovcakova L, Hao-Shen H, et al. (2015) Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood. 125: 2131-2140. [PubMed.]
- Mead AJ, Chowdhury O, Pecquet C, Dusa A, Woll P, et al. (2013) Impact of isolated germline JAK2V617I mutation on human hematopoiesis. Blood. 121: 4156-4165. [Ref.]
- Marty C, Saint-Martin C, Pecquet C, Grosjean S, Saliba J, et al. (2014) Germline JAK2 mutations in the kinase domain are responsible for hereditary thrombocytosis and are resistant to JAK2 and HSP90 inhibitors. Blood. 123: 1372-1383. [PubMed.]
- Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. (2015) The role of JAK/STAT signaling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 113(3): 365-371. [PubMed.]
- Bain BJ, Ahmad S. (2014) Should myeloid and lymphoid neoplasms with PCM1-JAK2 and other rearrangements of JAK2 be recognized as specific entities? Br J Haematol. 166: 809-817. [PubMed.]
- Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, et al. (2008) Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 205: 751-758. [PubMed.]
- Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, et al. (2011) FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 118(14): 3911-3921. [Ref.]
- Hughes TP, Hochhaus A, Branford S, Müller MC, Kaeda JS, et al. (2010) Long-term pro-gnostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood. 116: 3758-3765. [PubMed.]
- Rui L, Schmitz R, Ceribelli M, Staudt LM. (2011) Malignant pirates of the immune system. Nat Immunol. 12(10): 933-940. [PubMed.]
- Griffiths DS, Li J, Dawson MA, Trotter MW, Cheng YH, et al. (2011) LIF-independent JAK signaling to chromatin in embryonic stem cells uncovered from an adult stem cell disease. Nat Cell Biol. 13(1): 13-21. [PubMed.]
- Drennan AC, Rui L. (2017) HiJAKing the Epigenome in Leukemia and Lymphoma. Leuk Lymphoma. 58(11): 2540-2547. [PubMed.]
- Brooks AJ, Putoczki T. (2020) JAK-STAT Signaling Pathway in Cancer. Cancers (Basel). 12(7): 1971. [Ref.]
- Kan Z, Zheng H, Liu X, Li S, Barber TD, et al. (2013) Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 23: 1422-1433. [PubMed.]
- Kumar J, Fraser FW, Riley C, Ahmed N, McCulloch DR, et al. (2014) Granulocyte colony-stimulating factor receptor signaling via Janus kinase 2/signal transducer and activator of transcription 3 in ovarian cancer. Br J Cancer. 110: 133-145. [Ref.]
- He B, You L, Uematsu K, Zang K, Xu Z, et al. (2003) SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci USA. 100: 14133-14138. [Ref.]
- Duarte CW, Willey CW, Zhi D, Cui X, Harris JJ, et al. (2012) Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PloS One. 7(1): e29653. [PubMed.]
- Zalpoor H, Akbari A, Samei A, Forghaniesfidvajani R, Kamali M, et al. (2022) The roles of Eph receptors, neuropilin-1, P2X7, and CD147 in COVID-19- associated neurode-generative diseases: inflammasome and JaK inhibitors as potential promising therapies. Cell Mol Biol Lett. 27(1): 10. [Ref.]
- Brantley EC, Nabors LB, Gillespie GY, Choi Y-H, Palmer CA, et al. (2008) Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 14: 4694-4704. [PubMed.]
- Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D'Angeli L, et al. (2010) A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY). 2: 823-842. [PubMed.]
- Pawlus MR, Wang L, Hu CJ. (2014) STAT3 and HIF1 alpha cooperatively activate HIF1 target genes in MDAMB-231 and RCC4 cells. Oncogene. 33: 1670-1679. [PubMed.]
- Andersen MH. (2014) The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia. 28: 1784-1792. [PubMed.]
- Chun Pang, Yuan Gu, Yuechao Ding, Chao Ma, Wei Yv, et al. (2018) Several genes involved in the JAK-STAT pathway may act as prognostic markers in pancreatic cancer identified by microarray data analysis. Medicine (Baltimore). 97(50): e13297. [PubMed.]
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, et al. (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11: 1314-1321. [PubMed.]
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, et al. (2006) IL-2 regulates FOXP3expression in human CD4+ CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 108: 1571-1579. [PubMed.]
- Cho KH, Jeong KJ, Shin SC, Kang J, Park CG, et al. (2013) STAT3 mediates TGF-beta1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett. 336: 167-173. [PubMed.]
- Xie T-X, Wei D, Liu M, Gao AC, Ali-Osman F, et al. (2004) Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 23(20): 3550-3560. [PubMed.]
- Becker TM, Boyd SC, Mijatov B, Gowrishankar K, Snoyman S, et al. (2014) Mutant BRAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene. 33: 1158-1166. [PubMed.]
- Liu F, Cao J, Wu J, Sullivan K, Shen J, et al. (2013) Stat3-targeted therapies overcome the acquired resistance to vemurafenib in melanomas. J Invest Dermatol. 133: 2041-2049. [PubMed.]
- Qureshy Z, Johnson DE, Grandis JR. (2020) Targeting the JAK/STAT pathway in solid tumors. J Cancer Metastasis Treat. 6: 27. [PubMed.]
- Monnien F, Zaki H, Borg C, Mougin C, Bosset J-F, et al. (2010) Prognostic value of phosphorylated STAT3 in advanced rectal cancer: a study from 104 French patients included in the EORTC 22921 trial. J Clin Pathol. 63: 873-878. [PubMed.]
- Steinbuck MP, Winandy S. (2018) A Review of Notch Processing With New Insights Into Ligand- Independent Notch Signaling in T-Cells. Front Immunol. 9: 1230. [PubMed.]
- Martinez Arias A, Zecchini V, Brennan K. (2002) CSL independent Notch signaling: a checkpoint in cell fate decisions during development? Curr Opin Genet Dev. 12: 524-533. [PubMed.]
- Aster JC, Pear WS, Blacklow SC. (2017) The varied roles of notch in cancer. Annu Rev Pathol. 12: 245-275. [PubMed.]
- Nowell CS, Radtke F. (2017) Notch as a tumour suppressor. Nat Rev Cancer. 17: 145-159. [PubMed.]
- Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, et al. (2002) Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 8: 979-986. [PubMed.]
- Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, et al. (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic mediate tumorigenesis. Cancer Cell. 3: 565-576. [PubMed.]
- Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Olson S, et al. (2005) Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Res. 65: 3555-3561. [PubMed.]
- Konishi J, Yi F, Chen X, Vo H, Carbone DP, et al. (2010) Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene. 29: 589-596. [PubMed.]
- Sakamoto K, Ohara O, Takagi M, Takeda S, Katsube K. (2002) Intracellular cell autonomous association of Notch and its ligands: a novel mechanism of Notch signal modification. Dev Biol. 241: 313-326. [PubMed.]
- Yao K, Rizzo P, Rajan P, Albain K, Rychlik K, et al. (2011) Notch-1 and notch-4 receptors as prognostic markers in breast cancer. Int J Surg Pathol. 19: 607-613. [PubMed.]
- Kunanopparat A, Hirankarn N, Issara-Amphorn J, Tangkijvanich P, Sanpavat A. (2021) The expression profile of Jagged1and Delta-like 4 in hepatocellular carcinoma. Asian Pac J Allergy Immunol. 39(1): 44-52. [PubMed.]
- Xue TC, Zou JH, Chen RX, Cui JF, Tang ZY, et al. (2014) Spatial localization of the JAG1/ Notch1/osteopontin cascade modulates extrahepatic metastasis in hepatocellular carcinoma. Int J Oncol. 45: 1883-1890. [PubMed.]
- Yeh TS, Wu CW, Hsu KW, Liao WJ, Yang MC, et al. (2009) The activated Notch1 signal pathway is associated with gastric cancer progression through cyclooxygenase-2. Cancer Res. 69: 5039-5048. [PubMed.]
- Xiao HJ, Ji Q, Yang L, Li RT, Zhang C, et al. (2018) In vivo and in vitro effects of microRNA-124 on human gastric cancer by targeting JAG1 through the Notch signaling pathway, J Cell Biochem. 119: 2520-2534. [PubMed.]
- Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, et al. (2011) Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 71: 6061-6072. [PubMed.]
- Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG. (2013) Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res. 19: 3224-3233. [Ref.]
- Jubb AM, Browning L, Campo L, Turley H, Steers G, et al. (2012) Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology. 60:740-747. [PubMed.]
- Qiu XX, Chen L, Wang CH, Lin ZX, Chen BJ, et al. (2016) The vascular notch ligands delta-like ligand 4 (DLL4) and Jagged1 (JAG1) have opposing correlations with micro-vascularization but a uniform prognostic effect in primary glioblastoma: a preliminary study. World Neurosurg. 88: 447-458. [PubMed.]
- Qiu XX, Wang CH, You N, Chen BJ, Wang XF, et al. (2015) Jagged1 expression is associated with poor outcome in primary glioblastoma. Med Oncol. 32: 341. [PubMed.]
- Kim EJ, Kim SO, Jin X, Ham SW, Kim J, et al. (2015) Epidermal growth factor receptor variant III renders glioma cancer cells less differentiated by JAGGED1. Tumour Biol. 36: 2921-2928. [PubMed.]
- Yi L, Zhou X, Li T, Liu P, Hai L, et al. (2019) Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J Exp Clin Cancer Res. 38: 339. [Ref.]
- Giachino C, Boulay JL, Ivanek R, Alvarado A, Tostado C, et al. (2015) A tumor suppressor function for notch signaling in forebrain tumor subtypes. Cancer Cell. 28: 730-742. [PubMed.]
- Parmigiani E, Taylor V, Giachino C. (2020) Oncogenic and tumor-suppressive functions of NOTCH signaling in glioma. Cells. 9(10): 2304. [PubMed.]
- Xue S, He L, Zhang X, Zhou J, Li F, et al. (2017) Expression of Jagged1/Notch3 signaling pathway and their relationship with the tumor angiogenesis in TNBC. Arch Med Res. 48: 169-179. [PubMed.]
- Liu H, Wang J, Zhang M, Xuan Q, Wang Z, et al. (2017) Jagged1 promotes aromatase inhibitor resistance by modulating tumor-associated macrophage differentiation in breast cancer patients. Breast Cancer Res Treat. 166: 95-107. [PubMed.]
- Simoes BM, O’ Brien CS, Eyre R, Silva A, Yu L, et al. (2015) Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. 12: 1968-1977. [PubMed.]
- Cabrera RM, Mao SPH, Surve CR, Condeelis JS, Segall JE. (2018) A novel neuregulin jagged1 paracrine loop in breast cancer trans endothelial migration. Breast Cancer Res. 20: 24. [PubMed.]
- Strell C, Paulsson J, Jin SB, Tobin NP, Mezheyeuski A, et al. (2019) Impact of epithelial-stromal interactions on peritumoral fibroblasts in ductal carcinoma in situ. J Natl Cancer Inst. 111: 983-995. [PubMed.]
- Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, et al. (2014) Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS One. 9: e112424. [PubMed.]
- Wu C, Chen M, Sun Z, Ye Y, Han X, et al. (2019) Wenshen Zhuanggu formula mitigates breast cancer bone metastasis through the signaling crosstalk among the Jagged1/ Notch, TGF-beta and IL-6 signaling pathways. J Ethnopharmacol. 232: 145-154. [PubMed.]
- Sethi N, Dai X, Winter CG, Kang Y. (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 19: 192-205. [PubMed.]
- Parida S, Wu S, Siddharth S, Wang G, Muniraj N, et al. (2021) A procarcinogenic colon microbe promotes breast tumorigenesis and metastatic progression and concomitantly activates Notch and β-Catenin Axes. Cancer Discov. 11: 1138-1157. [PubMed.]
- Nabet BY, Qiu Y, Shabason JE, Wu TJ, Yoon T, et al. (2017) Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell. 170: 352-366. e313. [PubMed.]
- Krishna BM, Jana S, Singhal J, Horne D, Awasthi S, et al. (2019) Notch signaling in breast cancer: from pathway analysis to therapy. Cancer Lett. 461: 123-131. [PubMed.]
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, et al. (2005) High-level co-expression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor over-all survival. Cancer Res. 65: 8530-8537. [PubMed.]
- Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, et al. (2007) MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 39: 759-769. [PubMed.]
- Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, et al. (2016) HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 537: 102-106. [PubMed.]
- Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, et al. (2008) NUMB controls p53 tumour suppressor activity. Nature. 451: 76-80. [PubMed.]
- Dittmer J. (2018) Breast cancer stem cells: features, key drivers and treatment options. Semin Cancer Biol. 53: 59-74. [PubMed.]
- Ibrahim, SA, Gadalla R, El-Ghonaimy EA, Samir O, Mohamed HT, et al. (2017) Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL- 6/STAT3, Notch and EGFR signaling pathways. Mol Cancer. 16: 57. [PubMed.]
- Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, et al. (2017) Notch shapes the innate immunophenotype in breast cancer. Cancer Discov. 7: 1320-1335. [PubMed.]
- Groeneweg JW, Foster R, Growdon WB, Verheijen RH, Rueda BR. (2014) Notch signaling in serous ovarian cancer. J Ovarian Res. 7: 95. [PubMed.]
- Park JT, Chen X, Tropè CG, Davidson B, Shih I-M, et al. (2010) Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am J Pathol. 177: 1087-1094. [PubMed.]
- Choi JH, Park JT, Davidson B, Morin PJ, Shih I-M, et al. (2008) Jagged-1 and Notch3 juxtacrine loop regulates ovarian tumor growth and adhesion. Cancer Res. 68: 5716-5723. [Ref.]
- Hu W, Lu C, Dong HH, Huang J, Shen D-Y, et al. (2011) Biological roles of the Delta family Notch ligand Dll4 in tumor and endothelial cells in ovarian cancer. Cancer Res. 71: 6030-6039. [Ref.]
- Liu MX, Siu MK, Liu SS, Yam JW, Ngan HY, et al. (2014) Epigenetic silencing of micro-RNA-199b-5p is associated with acquired chemoresistance via activation of JAG1-Notch1 signaling in ovarian cancer. Oncotarget. 5: 944-958. [Ref.]
- Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, et al. (1996) Exclusive development of Tcell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 183: 2283-2291. [PubMed.]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, et al. (2014) ANOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 20: 1130-1137. [PubMed.]
- Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, et al. (2015) Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 21: 1182-1189. [PubMed.]
- Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, et al. (2014) Genomewide mapping and characterization f Notch regulated long noncoding RNAs in acute leukemia. Cell. 158: 593-606. [PubMed.]
- Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, et al. (2009) Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 113: 1689-1698. [PubMed.]
- Bernasconi-Elias P, Hu T, Jenkins D, Firestone B, Gans S, et al. (2016) Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene. 35: 6077-6086. [PubMed.]
- Tardivon D, Antoszewski M, Zanggern N, Nkosi M, Sordet-Dessimoz J, et al. (2012) Notch signaling promotes disease initiation and progression in murine chronic lymphocytic leukemia. Blood. 137: 3079-3092. [PubMed.]
- Huang Y-H, Cai K, Xu P-P, Wang L, Huang C-X, et al. (2021) CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7- NOTCH-CCL2/CSF1 axis. Signal Transduct Target Ther. 6: 10. [PubMed.]
- Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, et al. (2013) Notch activation inhibits AMLgrowth and survival: a potential therapeutic approach. J Exp Med. 210: 321-337. [PubMed.]
- Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP. (2016) Protein kinase Cι drives a NOTCH3-dependent Stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell. 29: 367-378. [Ref.]
- Sinicropi-Yao SL, Amann JM, Lopez DLY, Cerciello F,Coombes KR, et al. (2019) Co-expression analysis reveals mechanisms underlying the varied roles of NOTCH1 in NSCLC. J Thorac Oncol. 14: 223-236. [PubMed.]
- Hayakawa Y, Tsuboi M, AsfahaS, Kinoshita H, Niikura R, et al. (2019) BHLHA15-positive secretory precursor cells can give rise to tumors in intestine and colon in mice. Gastroenterology. 156: 1066-1081. e1016. [PubMed.]
- Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, O Lohuis J, et al. (2019) Epithelial NOTCH signaling rewires the tumor micro-environment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell. 36: 319-336. e317. [PubMed.]
- Bu P, Wang L, Chen K-Y, Srinivasan T, Murthy Lakshmi narasimha KP, et al. (2016) A miR-34a-numb feedforward loop triggered by inflammation regulates asymmetric stem cell division in intestine and colon cancer. Cell Stem Cell. 18: 189-202. [PubMed.]
- Ruland J. (2019) Colon cancer: epithelial notch signaling recruits neutrophils to drive metastasis. Cancer Cell. 36: 213-214. [PubMed.]
- Sonoshita M, Itatani Y, Kakizaki F, Sakimura K, Terashima T, et al. (2015) Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHO-GEF protein TRIO. Cancer Discov. 5: 198-211. [PubMed.]
- Jackstadt R, Sansom OJ. (2016) Mouse models of intestinal cancer. J Pathol. 238: 141-151. [PubMed.]
- Kranenburg O. (2015) Pro-metastatic NOTCH signaling in colon cancer. Cancer Discov. 5: 115-117. [PubMed.]
- Chanrion M, Kuperstein I, Barrière C, El Marjou F, Cohen D, et al. (2014) Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat Commun. 5: 5005. [PubMed.]
- Zhu C, Ho Y-J, Salomao MA, Dapito DH, Bartolome A, et al. (2021) Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J Hepatol. 74: 613-626. [PubMed.]
- Razumilava N, Gores GJ. (2013) Notch-driven carcinogenesis: the merging of hepatocellular cancer and cholangiocarcinoma into a common molecular liver cancer subtype. J Hepatol. 58: 1244-1245. [PubMed.]
- Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, et al. (2012) Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 143: 1660-1669. e1667. [PubMed.]
- Zhang L, Chen J, Yong J, Qiao L, Xu L, et al. (2019) An essential role of RNF187 in Notch 1 mediated metastasis of hepatocellular carcinoma. J Exp Clin Cancer Res. 38: 384. [Ref.]
- Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, et al. (2011) Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 208: 1963-1976. [PubMed.]
- Ferrarotto R, Mitani Y, McGrail DJ, Li K, Karpinets TV, et al. (2021) Proteogenomic analysis of salivary adenoid cystic carcinomas defines molecular subtypes and identifies therapeutic targets. Clin Cancer Res. 27: 852-864. [PubMed.]
- Oronsky B, Ma PC, Morgensztern D, Carter CA. (2017) Nothing But NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 19: 991-1002. [Ref.]
- Hu J, Wang Y, Zhang Y, Yu Y, Chen H, et al. (2019) Comprehensive genomic profiling of small cell lung cancer in Chinese patients and the implications for therapeutic potential. Cancer Med. 8: 4338-4347. [PubMed.]
- Quintanal-Villalonga A, Taniguchi H, Zhan YA, Hasan MM, Chavan SS, et al. (2021) Multiomic analysis of lung tumors defines pathways activated in neuroendocrine transformation. Cancer Discov. 11: 3028-3047. [PubMed.]
- Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, et al. (2015) A DLL3-targeted antibody-drug conjugate eradicates high grade pulmonary neuroendocrine tumorinitiating cells in vivo. Sci Transl Med. 7: 302ra136. [PubMed.]
- Xie H, Boland JM, Maleszewski JJ, Aubry MC, Yi ES, et al. (2019) Expression of delta like protein 3 is reproducibly present in a subset of small cell lung carcinomas and pulmonary carcinoid tumors. Lung Cancer. 135: 73-79. [Ref.]
- Gazdar AF, Bunn PA, Minna JD. (2017) Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 17: 725-737. [PubMed.]
- George J, Lim JS, Jang SJ, Cun Y, Ozretić L, et al. (2015) Comprehensive genomic profiles of small cell lung cancer. Nature. 524: 47-53. [PubMed.]
- Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. (2006) Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem. 281: 39819-39830. [PubMed.]
- Wang H, Chen Y, Fernandez-Del Castillo C, Yilmaz O, Deshpande V. (2013) Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: a critical look at notch signaling pathway. Mod Pathol. 26: 139-147. [PubMed.]
- Rekhtman N, Pietanza MC, Hellmann MD, Naidoo J, Arora A, et al. (2016) Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin Cancer Res. 22: 3618-3629. [PubMed.]
- Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, et al. (2016) ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 16: 1259-1272. [PubMed.]
- Meder L, König K, Ozretić L, Schultheis AM, Ueckeroth F, et al. (2016) NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer. 138: 927-938. [PubMed.]
- Wyche TP, Dammalapati A, Cho H, Harrison AD, Kwon GS, et al. (2014) Thiocoraline activates the Notch pathway in carcinoids and reduces tumor progression in vivo. Cancer Gene Ther. 21: 518-525. [PubMed.]
- Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, et al. (2019) Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell. 179: 403-416. e23. [PubMed.]
- Quan XX, Hawk NV, Chen W, Coupar J, Lee SK, et al. (2018) Targeting Notch1 and IKKα enhanced NF-κB activation in CD133(+) skin cancer stem cells. Mol Cancer Ther. 17: 2034-2048. [PubMed.]
- Alderuccio JP, Lossos IS. (2022) NOTCH signaling in the pathogenesis of splenic marginal zone lymphoma-opportunities for therapy. Leuk Lymphoma. 63(2): 279-290. [PubMed.]
- Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, et al. (1999) Notch signaling controls pancreatic cell differentiation. Nature. 400: 877-881. [PubMed.]
- Oishi H, Sunamura M, Egawa S, Motoi F, Unno M, et al. (2010) Blockade of delta-like ligand 4 signaling inhibits both growth and angiogenesis of pancreatic cancer. Pancreas. 39: 897-903. [PubMed.]
- Mazur PK, Einwächter H, Lee M, Sipos B, Nakhai H, et al. (2010) Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 107: 13438-13443. [PubMed.]
- Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, et al. (2012) The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther. 11: 1999-2009. [PubMed.]
- Hu Y, Su H, Li X, Guo G, Cheng L, et al. (2015) The NOTCH ligand JAG GED2 promotes pancreatic cancer metastasis independent of NOTCH signaling activation. Mol Cancer Ther. 14: 289-297. [PubMed.]
- Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, et al. (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 3: 565-576. [PubMed.]
- Thomas MM, Zhang Y, Mathew E, Kane KT, Maillard I, et al. (2014) Epithelial Notch signaling is a limiting step for pancreatic carcinogenesis. BMC Cancer. 14: 862. [Ref.]
- Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, et al. (2011) Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer. 10: 138. [Ref.]
- Gao J, Long B and Wang Z. (2017) Role of Notch signaling pathway in pancreatic cancer. Am J Cancer Res. 7: 173-186. [Ref.]
- Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, et al. (2010) Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 101: 293-299. [PubMed.]
- Raimondi C, Gradilone A, Naso G, Vincenzi B, Petracca A, et al. (2011) Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res Treat. 130: 449-455. [PubMed.]
- Shao S, Zhao X, Zhang X, Luo M, Zuo X, et al. (2015) Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol Cancer. 14: 28. [Ref.]
- Lu J, Ye X, Fan F, Xia L, Bhattacharya R, et al. (2013) Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 23: 171-185. [PubMed.]
- Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, et al. (2017) Endothelial Notch 1 Activity Facilitates Metastasis. Cancer Cell. 31: 355-367. [PubMed.]
- Schmalhofer O, Brabletz S, Brabletz T. (2009) E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 28: 151-166. [PubMed.]